Aicardi-Gouti-Kurrres-syndrom: fænotypisk og genetisk spektrum i en serie på tre tilfælde | Anales de Pediatr Larra
Aicardi-Gouti-Kurrres-syndrom (AGS) er en sjælden arvelig sygdom, hvis nøjagtige prævalens er ukendt. Det blev først beskrevet i 1984 af Jean Aicardi og Francoise Gouti Larres som en progressiv encefalopati med debut i de første måneder af livet karakteriseret ved cerebrospinalvæske (CSF) lymfocytose og forkalkninger i de basale ganglier.1 det manifesterer sig med irritabilitet, psykomotorisk retardation, spasticitet, dystoni, epileptiske anfald, tilbagevendende episoder med aseptisk feber og mikrocefali. Dødeligheden er højere i den encephalopatiske fase, og selvom sygdommen typisk stabiliseres bagefter, forårsager den alvorlige neurologiske følger. Andre karakteristiske træk, der kan forekomme i løbet af kurset, er chilblains, okulære symptomer (hovedsageligt glaukom), hjerteinddragelse eller autoimmune lidelser.2 type i interferoner spiller en afgørende rolle i patogenesen af AGS, hvor deres ekspression opreguleres, hvilket fører til øget produktion.3 af denne grund er et af de klassiske laboratoriefund hos disse patienter et forhøjet niveau af interferon alfa i CSF sammen med pleocytose og lige så forhøjede niveauer af neopterin og biopterin. Den potentielle anvendelighed ved at vurdere niveauet af ekspression af interferon-stimulerede gener af interferon i perifert blod som markør undersøges i øjeblikket, da der er tegn på, at disse niveauer forbliver høje forbi den encephalopatiske fase (“interferon signatur”).3-5 en anden nøglefunktion er påvisning af neuroimaging abnormiteter inklusive forkalkninger i de basale ganglier og ændringer i det hvide stof (Fig. 1). Hidtil kender vi 7 gener, hvis mutationer kan føre til opregulering af interferonvejen: ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREKS1 og IFIH1. Der er beskrevet heterogene mutationer for TREKS1 -, ADAR-og IFIH1-generne, hvorimod mutationerne rapporteret i alle andre gener har været homogene.2 mutationer i IFIH1-genet blev påvist Senest (2014) 4 og er derfor de mindst hyppige patogene varianter, mens mutationer i RNASEH2B-og TREKS1-generne tegner sig for den højeste andel af diagnosticerede tilfælde af AGS.
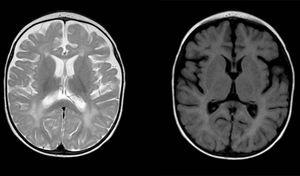
Diffuse og ujævn signalabnormiteter i hvidt stof i begge hjernehalvdel, hyperintense i T2-vægtede billeder. Forstørret subarachnoid rum med frontotemporal overvejelse i begge halvkugler, med udvidelse af den interhemisfæriske fissur og øget ventrikulær størrelse (i fravær af øget tryk), kompatibel med kortikal og subkortisk atrofi.
i de sidste par årtier, takket være fremskridt inden for genetik, der tillader påvisning af disse specifikke mutationer, er der fremkommet bevis for et bredt fænotypisk spektrum ud over den klassiske præsentation baseret på det forårsagende gen. Vi præsenterer tilfælde af 3 patienter, der har fået en diagnose af AGS i de sidste 8 år med det formål at analysere deres kliniske træk i forhold til den underliggende genetiske defekt (tabel 1). Generelt var de præsenterende træk ved AGS i overensstemmelse med dem, der er beskrevet i den seneste sagsserie i litteraturen: neonatal præsentation (33%), mikrocefali (66%), psykomotorisk retardation (100%), spasticitet (100%), alvorlig intellektuel handicap (66%) og forkalkninger på kranial CT (66%), skønt kun en patient havde epileptiske anfald.
Karakteristik af patienter med Aicardi-Gouti Kurrres syndrom.
| sag 1 | sag 2 | sag 3 | |
|---|---|---|---|
| genetik | homosygmutation (P. Ala177Thr) i RNASEH2B-gen | Homosygmutation (341g>a) i TREKS1-gen | Heterosygmutation (c.992c>g og p.Thr331Arg) i IFIH1 gen |
| nuværende alder | 3 år | 7 år og 4 måneder | 12 år og 11 måneder |
| køn | mand | kvinde | mand |
| Oprindelse | Rumænien | Spanien | Italien |
| AP | – | uge 36: intrauterin vækstbegrænsning uge 37: mikrocefali, placental forkalkning |
ganespalte |
| kliniske manifestationer | |||
| alder ved debut | 10 måneder | fødsel | 2 år |
| indledende præsentation | irritabilitet psykomotorisk regression |
tremor, hypotoni, svag gråd, vækstfejl | motorisk færdighedsforsinkelse |
| psykomotorisk retardering | Ja | Ja | Ja |
| sprog | 2-stavelse words | No | Yes |
| Microcephaly | Yes | Yes | No |
| Chilblains | No | Yes | Yes |
| Epilepsy | No | Yes | No |
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFCS IV |
| bevægelsesforstyrrelse | Nej | Ja | Nej |
| unormale øjenbevægelser | Nej | Nej | Nej |
| synshandicap | Nej | – | nærsynethed |
| glaukom | Nej | Nej | Nej |
| høretab | – | – | Nej |
| hjerteinddragelse | Nej | Mild tricuspid og mitral regurgitation | Nej |
| tilbagevendende feber | Nej | Nej | Nej |
| intellektuel handicap | Ja | Ja, grav | Ja, mild |
| andet | – | – | Singleton-Merten syndrom: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
| CSF | |||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – |
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – |
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – |
| INF alfa (IU/mL) | 25 ( | 75 ( | – |
| Blood | |||
| Interferon signature | – | – | Gene overexpression |
| Neuroimaging | |||
| Calcifications on CT | No | Yes. I basale og periventrikulære ganglier | Ja. Symmetriske forkalkninger i dyb masseødelæggelsesvåben i frontal region og lentiform kerne |
| hoved MR | Diffuse og ujævn ændringer i masseødelæggelsesvåben intensitet af begge cerebrale halvkugler, hyperintense på T2. Involvering af subkortikale masseødelæggelsesvåben (skåner u-fibrene) og periventrikulære masseødelæggelsesvåben | generaliseret masseødelæggelsesvåben med overvægt af lobar masseødelæggelsesvåben inklusive de subkortikale u-fibre i frontale, temporale og occipitale lapper, bilateralt og symmetrisk, uden kortikal involvering | – |
CSF, cerebrospinalvæske; CT, computertomografi; GMFCS, grovmotorisk funktion klassificeringssystem; INF, interferon; IUGR, intrauterin vækstbegrænsning; MR, magnetisk resonansbilleddannelse; PNP, polyneuropati; masseødelæggelsesvåben, hvid mater.
som tidligere nævnt er homosygøse mutationer i RNASEH2B-genet de hyppigste varianter, der forårsager AGS, og deres fænotypiske ekspression svarer normalt mest til den klassiske præsentation.4 Dette var tilfældet med patienten i vores undersøgelse, der bar en sådan mutation, der var begyndt i en alder af 10 måneder med irritabilitet og psykomotorisk retardation og med karakteristiske neuroimaging-og CSF-fund.
tyve procent af tilfælde af AGS kan have en neonatal præsentation med sygdomsudbrud, der forekommer i utero.5 mutationer i et hvilket som helst af de 7 førnævnte gener kan føre til denne fænotype, men denne tidlige præsentation er hyppigst forbundet med Treks-genet.4,5 den indledende præsentation af denne form svarer til præsentationen af en TORCH-infektion med hepatosplenomegali, hypertransaminasæmi, trombocytopeni og neurologiske manifestationer inklusive ekstrem irritabilitet, bevægelsesforstyrrelser og epileptiske anfald.5 disse patienter har et mere alvorligt sygdomsforløb og har højere risiko for død. Patienten i vores prøve, der præsenterede med en sådan variant, havde en neonatal præsentation og har i øjeblikket den mest alvorlige form for sygdom i 3.
mutationer i ADAR1-genet og især ifih1-genet er forbundet med en sen begyndelse af symptomer efter 1 års levetid med normal psykomotorisk udvikling.5 i nogle af disse tilfælde har syndromet et godartet kursus med relativ bevarelse af sprog og motoriske færdigheder. Vores patient med en mutation i ifih1-genet var et enestående tilfælde, idet han også havde Singleton-Merten syndrom, en sjælden sygdom også forårsaget af en mutation i ifih1-genet og karakteriseret ved dental dysplasi, aorta forkalkninger og osteoporose.6
vores mål er at understrege den signifikante fænotypiske variabilitet af AGS og dens tilknytning til specifikke mutationer med det formål både at tilskynde til overvejelse af denne diagnose i tilfælde med præsentationer, der afviger fra den klassiske sygdomsform og at bidrage med yderligere information om sygdomsforløbet og resultaterne hos disse patienter.