Aicardi-Goutières syndrom: Fenotypové a genetické spektrum v sérii tří případů | Anales de Pediatría
Aicardi-Goutières syndrom (AGS) je vzácné dědičné onemocnění, jehož přesná prevalence není známa. To byla poprvé popsána v roce 1984 Jean Aicardi a Francoise Goutières jako progresivní encefalopatie s nástupem v prvních měsících života se vyznačuje mozkomíšního moku (CSF), lymfocytóza a kalcifikace v bazálních gangliích.1 projevuje se podrážděností, psychomotorickou retardací, spasticitou, dystonií, epileptickými záchvaty, recidivujícími epizodami aseptické horečky a mikrocefalií. Úmrtnost je vyšší během encefalopatické fáze, a ačkoli se nemoc obvykle stabilizuje poté, způsobuje závažné neurologické následky. Dalšími charakteristickými rysy, které se mohou objevit během jeho průběhu, jsou chilblains, oční příznaky (zejména glaukom), srdeční postižení nebo autoimunitní poruchy.2 interferony typu I hrají klíčovou roli v patogenezi AGS, ve které je jejich exprese upregulována, což vede ke zvýšené produkci.3 z tohoto důvodu, jeden z klasických laboratorních nálezů u těchto pacientů je zvýšené hladiny interferonu alfa v CSF, spolu s pleocytózou a stejně tak zvýšené hladiny neopterin a biopterin. Potenciální užitečnost hodnocení úrovně exprese interferonem stimulovaných genů pomocí interferonu v periferní krvi jako markeru je v současné době vyšetřován, protože tam je důkaz, že tyto úrovně zůstat vysoká kolem encefalopatických fáze („interferon podpis“).3-5 dalším klíčovým rysem je detekce neuroimagingových abnormalit včetně kalcifikací v bazálních gangliích a změn v bílé hmotě (obr. 1). K dnešnímu dni, víme, že 7 genů, jejichž mutace může vést k upregulace interferon cesty: ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1 a IFIH1. Heterozygotní mutace byly popsány pro geny TREX1, ADAR a IFIH1, zatímco mutace hlášené ve všech ostatních genech byly homozygotní.2 mutace v genu IFIH1 byly detekovány Naposledy (2014)4 a jsou proto nejméně častými patogenními variantami, zatímco mutace v genech RNASEH2B a TREX1 představují nejvyšší podíl diagnostikovaných případů AGS.
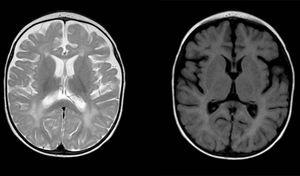
difúzní a nepravidelné signální abnormality v bílé hmotě v obou mozkových hemisférách, hyperintenze v obrazech vážených T2. Rozšířené subarachnoidálního prostoru s frontotemporální převaha v obou hemisfér, s rozšířením interhemisférické praskliny a zvýšení ventrikulární velikost (v nepřítomnosti zvýšeného tlaku), kompatibilní s kortikální a subkortikální atrofie.
V posledních několika desetiletích se díky pokroku v genetice, který umožňuje detekci těchto specifických mutací, důkazy se objevily široké fenotypové spektrum mimo klasické prezentace založené na kauzální gen. Představujeme případy 3 pacientů s diagnózou AGS za posledních 8 let s cílem analyzovat jejich klinické rysy ve vztahu k základní genetické vadě (Tabulka 1). Obecně platí, prezentující funkce AGS byly konzistentní s těmi, které popsal v nejnovějším případě série v literatuře: novorozenecké prezentace (33%), mikrocefalie (66%), psychomotorická retardace (100%), spasticity (100%), těžkým mentálním postižením (66%) a kalcifikace na kraniální CT (66%), i když pouze jeden pacient měl epileptický záchvat.
charakteristika pacientů se syndromem Aicardi-Goutières.
| Případ 1 | 2 | Případ 3 | |
|---|---|---|---|
| Genetika | Homozygotní mutace (p.Ala177Thr) v RNASEH2B gen | Homozygotní mutace (341G>A) v TREX1 gen | Heterozygotní mutace (c.992C>G a p.Thr331Arg) v IFIH1 genu |
| Aktuální věk | 3 roky | 7 let a 4 měsíce | 12 let a 11 měsíců |
| Sex | Muž | Žena | Muž |
| Původ | Rumunsko | Španělsko | Itálie |
| AP | – | Týden 36: nitroděložní omezení růstu Týden 37: mikrocefalie, kalcifikace placenty |
Rozštěp patra |
| Klinické projevy | |||
| Věk při začátku | 10 měsíců | Narození | 2 roky |
| Úvodní prezentace | Podrážděnost Psychomotorický regrese |
Třes, hypotonie, slabý pláč, porucha růstu | Motorické zpoždění |
| Psychomotorické retardace | Ano | Ano | Ano |
| Jazyk | 2 Slabiky words | No | Yes |
| Microcephaly | Yes | Yes | No |
| Chilblains | No | Yes | Yes |
| Epilepsy | No | Yes | No |
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFCS IV |
| porucha Pohybu | Ne | Ano | Ne |
| Abnormální pohyby očí | Ne | Ne | Ne |
| Zrakové postižení | Ne | – | Krátkozrakost |
| Glaukom | Ne | Ne | Ne |
| ztráta Sluchu | – | – | Ne |
| Srdeční zapojení | Ne | Mírné trikuspidální a mitrální regurgitace | Ne |
| Opakující se horečka | Ne | Ne | Ne |
| mentální postižení | Ano | Ano, hrob | Ano, mírné |
| Další | – | – | Singleton-Merten syndrom: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
| CSF | |||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – |
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – |
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – |
| INF alfa (IU/mL) | 25 ( | 75 ( | – |
| Blood | |||
| Interferon signature | – | – | Gene overexpression |
| Neuroimaging | |||
| Calcifications on CT | No | Yes. V bazálních a periventrikulárních gangliích | Ano. Symetrické kalcifikace v hluboké WM čelní oblasti a lentiform jádro |
| MR Hlavy | Difuzní a nerovnoměrné změny ve WM intenzita obou mozkových hemisfér, hyperintenzivní v T2. Zapojení subkortikálních WM (šetřící U vláken) a periventrikulární WM | Všeobecné WM zapojení s převahou lobární WM včetně podkorových U vláken čelní, spánkové a týlní laloky, bilaterálně a symetricky, bez kortikální zapojení | – |
CSF, mozkomíšního moku; CT, počítačová tomografie; GMFCS, Gross Motor Function Classification System; INF, interferon; IUGR, nitroděložní omezení růstu; MRI, magnetic resonance imaging; PNP, polyneuropatie; WM, bílá mater.
Jak již bylo uvedeno dříve, homozygotní mutace v RNASEH2B genu jsou nejčastější varianty, které způsobují AGS a jejich fenotypové vyjádření obvykle odpovídá nejvíce klasické prezentaci.4 To byl případ pacienta v naší studii, která provádí takové mutace, která měla nástup ve věku 10 měsíců s podrážděnost a psychomotorické retardace a s charakteristickou neuroimaging a CSF zjištění.
dvacet procent případů AGS může mít novorozeneckou prezentaci s nástupem onemocnění vyskytujícím se in utero.5 mutací v kterémkoli ze 7 výše uvedených genů může vést k tomuto fenotypu, ale tato časná prezentace je nejčastěji spojována s genem TREX.4,5 úvodní prezentaci této formy je podobná jako POCHODEŇ infekce, s hepatosplenomegalie, hypertransaminasaemia, trombocytopenie a neurologických projevů včetně extrémní podrážděnost, poruchy hybnosti a epileptické záchvaty.5 tito pacienti mají závažnější průběh onemocnění a jsou vystaveni vyššímu riziku úmrtí. Pacient v našem vzorku, který předložil takovou variantu, měl novorozeneckou prezentaci a v současné době má nejzávažnější formu nemoci 3.
mutace v genu ADAR1 a zejména genu IFIH1 jsou spojeny s pozdním nástupem příznaků po 1 roce života s normálním psychomotorickým vývojem.5 v některých z těchto případů má syndrom benigní průběh s relativním zachováním jazykových a motorických dovedností. Náš pacient s mutací v IFIH1 genu byla singulární v případě, že on také měl Singleton-Merten syndrom, vzácné onemocnění také způsobena mutací v IFIH1 genu a vyznačuje dentální dysplazie, aortální kalcifikace a osteoporózy.6
Naším cílem je podtrhnout významné fenotypové variability AGS a jeho spojení s konkrétní mutací za účelem povzbudivé zvážení této diagnózy v případech s prezentací, které se odchylují od klasické formy onemocnění a přispět další informace o průběhu onemocnění a výsledků u těchto pacientů.