Gynekologie A Porodnictví kazuistika
Klíčová slova
Tetra-amelia; Malformace; Genotyp; Fenotyp.
Úvod
anomálie končetin představují důležitou skupinu vrozených patologií charakterizovaných hypoplazií nebo úplnou aplazií jedné nebo více kostí končetin. Abnormality končetin všech typů se vyskytují přibližně u 1 z 1300 až 2 000 narozených. Tyto anomálie končetin mohou být izolovány nebo spojeny s jinými anomáliemi . Tetraameliův syndrom je vzácný a šedé oblasti zůstávají.
zpráva o dvou případech tetra-amelia v úrovni II mateřství v Dakaru (Senegal) s podobné tetraamelia-1 (chromosom 17q21), tetraamelia-2 (chromozom 8q23) a Robert syndrom (chromozóm 8p21). To ilustruje obtížnost korelace fenotypu a zapojených genů.
Zprávy
1
Paní AD byl 44-rok-stará matka podle našeho oddělení v 36. týdnu těhotenství s těžkou preeklampsií a fetální anomálie. Bylo jí pět para bez anamnézy fetálních anomálií. Nyní nekouřila a nikdy nekouřila ani nepila alkohol. Nebyla vystavena pasivnímu kouření. Byla v příbuzenském manželství třetího stupně pro všechny své děti. Paní AD měla negativní test na hepatitidu B, HIV a syfilis. Byla chráněna před virem zarděnek a neměla předchozí expozici Toxoplasma gondii. Ultrazvukové monitorování prováděné pozdě 33 týdny a 35 týdny těhotenství získaly oligoamniózu a hydrocefalus, jakož i agenezi končetin. Recepty během těhotenství zahrnovaly podávání železa a kyseliny listové, stejně jako správa sulfadoxine pyrimethamine. Ten byl předepsán po 18 týdnech a poté 26 týdnech jako součást politiky profylaxe proti malárii pro těhotné ženy. Výška symphyseal-Fund měřila 28 cm. Kvůli závažným rysům preeklampsie; byla okamžitě hospitalizována a pozorována v porodním a porodním oddělení. Poté zpočátku dostávala IV síran hořečnatý k prevenci eklampsie a antihypertenziv k udržení systolického krevního tlaku pod 160 mmHg a diastolického krevního tlaku pod 105 mmHg.
bylo rozhodnuto o okamžitém císařském porodu. Bylo extrahováno 2 150 gramů živě narozených, které následně zemřely během 10 minut. Před smrtí tělo představovalo plazivé pohyby. Bylo identifikováno několik vnějších anomálií (Obrázek 1) včetně úplné ageneze všech čtyř končetin, hydrocefalus s obvodem hlavy 39 cm. Na obličeji byl hypertelorismus s kolobomem očních víček, mírným exoftalmem a aniridií. Ústa byla podobná obrácenému V bez jasného vymezení rtů a nos byl rudimentární. Novorozenec byl zbaven příkrovů (vlasů a obočí). Uši byly redukovány na náčrtky, které vypadaly jako štěrbiny. Krk byl krátký. Kmen byl redukován na 26 cm dlouhou kuželovitou strukturu s pupeční šňůrou na spodním konci. Hrudník byl úzký. Těsně pod pupkem, kufru rozšířil dozadu byl přítomen na podlaze kaudální pól, střední čáře s recesí a začínající což může odpovídat falus neurčeného typu. Byla zaznamenána ageneze pánve, genitálií a anální imperforace. Fetální patologie nebyla provedena. Smrt do 10 minut po porodu a kónický vzhled hrudníku však mohou naznačovat abnormality plic.
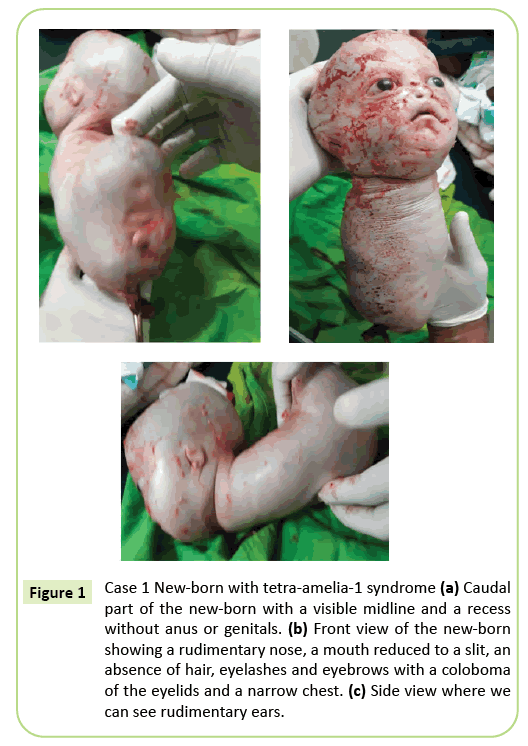
Obrázek 1: Případ 1 novorozenec se syndromem tetra-amelia-1 (a) kaudální část novorozence s viditelnou středovou linií a vybráním bez konečníku nebo genitálií. (b) Čelní pohled na nově narozené ukazuje primitivní nos, ústa snížena na štěrbiny, absence vlasy, řasy a obočí s kolobom víček a úzký hrudník. (c) boční pohled, kde můžeme vidět rudimentární uši.
Případ 2
druhý případ byl 22-letá primigravida podle našeho zařízení pro ultrazvukové skenování na 37 týdnů těhotenství. Nebyla v příbuzenském manželství. Měla negativní test na hepatitidu B, HIV a syfilis. Nebyla testována na toxoplazmózu a zarděnky. Během těhotenství nebylo provedeno žádné ultrazvukové sledování. Klinické vyšetření bylo v souladu s retardací růstu plodu (výška pozadí: 26 cm). Ultrazvukové nálezy ukázaly, že humerus je zkreslený měřením 23,9 mm, což odpovídá 17. týdnu těhotenství. Došlo k agenezi stehenní kosti. Iliakální křídla byla viditelná na ultrazvuku. Nebyly zjištěny žádné abnormality plic nebo srdce. Dodávka byla zahájena. Novorozenec měl ženský fenotyp s Apgarovým skóre 9 v 5. minutě. Morfologie hlavy a kmene byla bez zvláštnosti. Horní končetiny byly redukovány na dva 3 cm dlouhé pahýly. Byla zaznamenána kompletní ageneze 2 dolních končetin. Byla to symetrická anomálie (Obrázek 2).
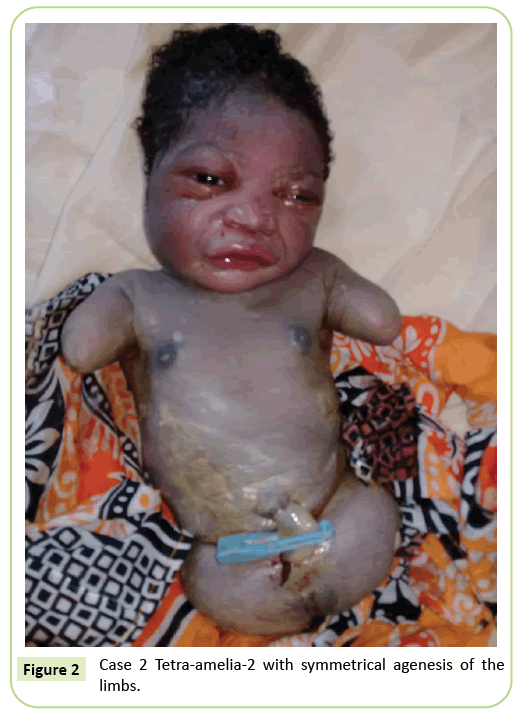
Obrázek 2: Případ 2 Tetra-amelia-2 se symetrickou agenezí končetin.
Diskuse
Bermejo-Sanchez et al. v roce 2011 byla popsána epidemiologie vrozené Amelie pomocí údajů shromážděných z 20 programů sledování vrozených anomálií ze všech kontinentů kromě Afriky v letech 1968 až 2006. Celkem bylo identifikováno 326 případů Amelie mezi 23 110 591 živě narozenými, mrtvě narozenými a potraty. Prevalence byla 1, 41 / 100 000 .
Tetra-amelia označuje jako úplnou nepřítomnost končetin a vyskytuje se vzácněji. Pokud je nám známo, tetra-amelia-1 je popsána v 7 rodinách. Zdá se, že následuje autozomálně recesivní dědičnost. Ve všech rodinách, tetra-amelia-1 byl spojován s těžkými malformacemi ostatních částí těla, včetně obličeje a hlavy, anomálie nervového systému, kostra, a genitálie. Plíce byly v mnoha případech nedostatečně rozvinuté, což ztěžuje nebo znemožňuje dýchání . Zimmer et al. v roce 1985 byla hlášena silně inbrední rodina, ve které 6 dětí mělo tetra-amelia-1 a hydrocefalus. V jednom z plodů popsali úplnou nepřítomnost pánevní kosti, rozštěpu rtu a patra, arrhinii a aplazii uší. Byly také zaznamenány bilaterální levé plíce, přetrvávající arteriální kanál, anální imperforace. Fetální testování eliminovalo diagnózu Robertova syndromu . Mezi další případy získané v literatuře patří případy Kosaki et al., v roce 1996 s plodem karyotypu 46, XX s tetrafokomelií a těžkou plicní hypoplazií kromě anomálií obličeje a hlavy . Rosenak a kol. popsal případ tetra-Amelie s těžkou plicní hypoplazií u dvou plodů nekrvavého páru. Fetální testování vyloučilo diagnózu Robertova syndromu . Dva další případy byly hlášeny Zlotogora et al. v roce 1993. Oba pacienti zemřeli brzy po narození a autoři navrhli existenci plicní hypoplasie. Niemann et al. hlásil příbuznou tureckou rodinu, ve které 4 z 8 bratrů trpěli tetra-Amelií. Kromě absence 4 končetin, vyšetření plodu ze 3 plodů odhalil několik anomálií: rozštěp rtů a /nebo palatine, laparoschisis, plicní anomálie, hypoplázie pánve, atrézie choanas, vagína a anální imperforation . Nakonec v roce 2005 Krahn et al. popsali 2 bratry narozené inbredním rodičům trpícím tetraamelií a těžkou plicní hypoplazií. Klíční kosti a lopatky byly u druhého plodu normální. Karyotyp byl normální .
Tetra-amelia-1 syndrom nebo TETAMS1 je způsobena homozygotní mutace v WNT3 gen na chromozomu 17q21 s autosomálně recesivní dědičnost. Syndrom Tetraamelia-2 (TETAMS2) je charakterizován rudimentárními končetinami nebo úplnou absencí končetin, obecně symetrickou i bilaterální agenezí plic v některých případech. Jsou také obvyklé anomálie plicního cévního systému a dysmorfie včetně bilaterálního rozštěpu rtu a patra, ankyloglossie, mandibulární hypoplazie, mikroretrognathie a labioskrotální aplazie .
Szenker-Ravi, studující 4 rodiny tetra-Amelie s agenezí nebo plicní hypoplazií, zaznamenal fenotypovou heterogenitu s anomáliemi končetin různé závažnosti . Sekvenování exomu v těchto 4 rodinách umožnilo identifikovat zkrácené homozygotní mutace v genu RSPO2 . Syndrom Tetraamelia-2 je způsoben homozygotní mutací v genu RSPO2 (610575) umístěném na chromozomu 8q23 .
fenotyp prvního případu popsaného v tomto článku odpovídá syndromu tetra-amelia-1 způsobenému zejména přítomností hydrocefalu, anomálií genitálií a rudimentárního nosu. Úzký hrudník a předčasná smrt před 10. minutou života naznačují těžkou plicní hypoplazii. Tento případ zdůrazňuje fenotypovou heterogenitu s kolobomem víček, hypertelorismem, exoftalmem a vzácnými přídavky.
druhý případ v naší studii považujeme za syndrom tetramelia-2 s ohledem na symetrickou tetra-Amelii s přítomností pahýlů horní končetiny. Diagnóza tetra-amelia by měla být provedena brzy během ultrazvukového sledování. Proto by mělo být zvýšeno povědomí o důležitosti ultrazvukového monitorování a použití 3D/4D ke zlepšení výsledků screeningu. Diagnóza pánevní hmoty na ultrazvuku spárované s Amelií by měla vyvolat podezření na syndrom defektu končetiny spleno-gonadální fúze.
kromě toho, vyšetření plodu a fetální testování pomocí rozvíjející technologie chromozomální microarray a exome a sekvenování genomu mají být podporována v naší nastavení. Lepší charakterizace případů umožňuje poskytovat rady párům a lepší znalost těchto klinických anomálií.
závěr
Tetra-ameliův syndrom je vzácný a šedé oblasti stále zůstávají. Tyto dva případy, ve srovnání s tím, co je již popsáno v literatuře, ilustrují fenotypovou heterogenitu tetraamelie. Vzhledem vzácný výskyt těchto anomálií, je důležité vytvořit mezinárodní registr anomálie, aby se zprávy o případech a nastavit vzorek banka pro rozšířené genetické studie k rodičům.
- Wilcox WR, Coulter CP, Schmitz ML (2015) vrozené poruchy deficitu končetin. Clin Perinatol 42: 281-300.
- Bermejo SE, Cuevas L, Amar E (2011) Amelia: multi-centrum popisné epidemiologické studie ve velké datový soubor z Mezinárodního Střediska pro vrozené Vady Dohled a Výzkum, a přehled literatury. Am J Med Genet C Semin Med Genet 157: 288-304.
- Zlotogora JSM, Shabany YO, Jarallah RY (1993) syndrom tetraamelie s plicní hypoplazií. Am J Med Genet 47: 570-571.
- Zimmer EZ (1985)Tetra-amelia s mnohočetnými malformacemi u šesti samčích plodů v jednom příbuzném. Europ. J Pediat 144: 412-414.
- Rosenak D (1991) recidivující tetraamelia a plicní hypoplazie s mnohočetnými malformacemi u sibirek. Am J Med Genet 38: 25-28.
- Gershoni BR (1990) Robertsův syndrom nebo „X-spojená amelia“? . Am J Med Genet 37: 569-572.
- Kosaki K, Jones MC, Stayboldt C (1996) Zimmer phocomelia: vymezení hlavní souřadnicové analýzy. Am J Med Genet 66: 55-59.
- Niemann S (2004) homozygotní mutace WNT3 způsobuje tetra-Amelii ve Velké příbuzenské rodině. Am J Hum Genet 74: 558-563.
- Krahn M (2005) Tetra-amelia a syndrom plicní aplazie: zpráva o nové rodině a vyloučení kandidátských genů. Clintonová 68: 558-560.
- Szenker-RE, Altunoglu U (2018) RSPO2 inhibice RNF43 a ZNRF3 řídí vývoj končetin nezávisle LGR4/5/6. Příroda 557: 564-569.