Pravidelné Paralyzuje
klinicky užitečné klasifikace primární periodické obrny, je uvedeno v Tabulce 1, zahrnuje hypokalemická, hyperkalemic, a paramyotonic formy.
Tabulka 1. Primární Periodická Paralýza (upraveno z Jurkat-Rott a Lehmann-Horn ) (Otevřít Tabulku v novém okně)
|
Onemocnění |
Gen |
Protein |
Dědictví |
Mutace |
|
HyperPP |
SCN4A |
Nav1.4 |
Dominantní |
v Pořádku |
|
NormoPP |
v Pořádku (ω-pórů) |
|||
|
Paramyotoniacongenita |
v Pořádku |
|||
|
HypoPP Typu II |
v Pořádku (ω-pórů) |
|||
|
HypoPP ABY |
CACNA1S |
Cav1.1 |
Dominantní |
Zisk (ω-pórů) |
|
ThyrotoxicPP |
KCNJ18 |
Kir2.18 |
Dominantní |
Ztráta |
|
Andersen-Tawil syndrom |
KCNJ2 |
Kir2.1 |
Dominantní |
Ztráta |
fyziologické základě ochablý slabina je inexcitability svalové membrány (tj. sarcolemma). Změna hladiny draslíku v séru není hlavní vadou primárního PP; změněný metabolismus draslíku je výsledkem PP. U primární a tyreotoxické PP dochází k ochablé paralýze s relativně malými změnami hladiny draslíku v séru, zatímco u sekundárního PP jsou hladiny draslíku v séru výrazně abnormální.
za tuto skupinu poruch není zodpovědný žádný jediný mechanismus. Jsou tedy heterogenní, ale sdílejí některé společné rysy. Slabost je obvykle zobecněna, ale může být lokalizována. Kraniální svalstvo a dýchací svaly jsou obvykle ušetřeny. Protahovací reflexy během útoků buď chybí, nebo se zmenšují. Svalová vlákna jsou během útoků elektricky nevyčerpatelná. Svalová síla je mezi útoky normální, ale po několika letech se u určitých typů PP (zejména primárního PP) vyvíjí určitý stupeň pevné slabosti. Všechny formy primárního PP (kromě Becker myotonia congenita) jsou buď autozomálně dominantní dědičné nebo sporadické (s největší pravděpodobností vznikající z bodových mutací).
napěťově citlivé iontové kanály úzce regulují generování akčních potenciálů (krátké a reverzibilní změny napětí buněčných membrán). Jedná se o selektivně a variabilně propustné iontové kanály. Energeticky závislé iontové transportéry udržují koncentrační gradienty. Během generování akčních potenciálů se ionty sodíku pohybují přes membránu přes napěťově řízené iontové kanály. Membrána klidových svalových vláken je polarizována primárně pohybem chloridu chloridovými kanály a je repolarizována pohybem draslíku. Kanály sodíku, chloridu a vápníkupatie jako skupina jsou spojeny s myotonií A PP. Funkční podjednotky sodíkových, vápníkových a draselných kanálů jsou homologní. Sodíkové kanálopatie jsou lépe pochopeny než kalciové nebo chloridové kanálopatie. Všechny formy familiárního PP ukazují konečnou mechanistickou dráhu zahrnující aberantní depolarizaci, inaktivaci sodíkových kanálů a nevyčerpatelnost svalových vláken.
Diskuse v tomto článku se primárně zabývá sodík, vápník, a draslík channelopathies, stejně jako sekundární formy PP. Chloridové kanálopatie nejsou spojeny s epizodickou slabostí a jsou podrobněji diskutovány v článcích o myotonických poruchách.
Shrnutí kanál dysfunkce v různých typech PP
S HyperPP rychlé inaktivace kanálu, mutace jsou obvykle nachází ve vnitřní části transmembránové segmenty nebo v intracelulárních smyček, které ovlivňují dokovací místa pro rychlé inaktivaci částic, čímž se zhoršuje rychle kanálu inaktivace vede k přetrvávající Na+ proudu.
S HypoPP hyperpolarizace-aktivní kation úniku působí proti K+ -opravuje aktuální, mutace způsobují nejvzdálenějších arginin nebo lysin substituce.
při úniku kationtů aktivovaných NormoPP depolarizací jsou mutace v hlubších lokalitách napěťového senzoru domény II na kodonu R675.
dysfunkce iontových kanálů je obvykle dobře kompenzována normální excitací a další spouštěče jsou často nezbytné k vytvoření svalové nevyčerpatelnosti v důsledku trvalé depolarizace membrány.
příjem glukózy a draslíku má u těchto poruch opačné účinky. V HyperPP vyvolává příjem draslíku útok, zatímco glukóza ho zlepšuje. Naproti tomu glukóza vyvolává hypokalemické záchvaty a draslík je léčbou útoku.
Všimněte si obrázku níže.
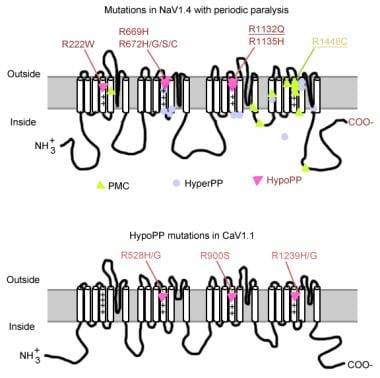 mutace v periodické paralýze.
mutace v periodické paralýze. svalový sodíkový kanál gen
sodíkový kanál má alfa podjednotku a beta podjednotku. Alfa podjednotka sodného kanálu je 260-kd glykoprotein obsahující asi 1800-2000 aminokyselin. Tento kanál je vysoce konzervovaný evolučně od Drosophily po člověka. Má 4 homologní domény (I-IV), které se skládají za vzniku centrálního póru, každý s 225-325 aminokyselinami. Každá doména se skládá ze 6 hydrofobních segmentů (S1-S6) procházejících buněčnou membránou. Mezi hlavní funkce kanálu patří napěťově citlivé bránění, inaktivace a selektivita iontů. Extracelulární smyčka mezi S5 a S6 namáčí do plazmatické membrány a podílí se na tvorbě pórů. Segment S4 obsahuje kladně nabité aminokyseliny v každé třetí poloze a funguje jako snímač napětí. Během depolarizace se mohou objevit změny konformace, což vede k aktivaci a inaktivaci kanálu. Buněčná smyčka mezi doménou III-S6 a doménou IV-S1 působí jako inaktivační brána.
sodíkový kanál má 2 brány (aktivace a inaktivace) a může existovat ve 3 stavech. V klidu s polarizovanou membránou se aktivační brána uzavře a otevře se inaktivační brána. Při depolarizaci se aktivační brána otevírá, což umožňuje iontům sodíku procházet iontovým kanálem a také vystavuje dokovací místo pro inaktivační bránu. Při pokračující depolarizaci se inaktivační brána uzavře, blokuje vstup sodíku do buňky a způsobuje vstup kanálu do rychle inaktivačního stavu. Tato inaktivace kanálu umožňuje repolarizaci membrány, což vede k návratu do klidového stavu se zavřenou aktivační bránou a otevřenou inaktivační bránou. V kosterním svalu savců se vyskytují dva inaktivační procesy: Rychlá inaktivace zahrnuje ukončení akčního potenciálu a působí v milisekundové Časové stupnici. Pomalá inaktivace trvá sekundy až minuty a může regulovat populaci excitovatelných sodíkových kanálů.
sodíkový kanál mutace, které narušují rychlé a pomalé inaktivace jsou obvykle spojeny s fenotyp HyperPP a myotonie, kde jako mutace, které zvyšují pomalu nebo rychle inaktivace produkovat ztráty sodíku kanálu funkce způsobit HypoPP.
mutace genu sodíkového kanálu (SCN4A) mají několik obecných rysů. Většina mutací jsou v „inaktivace“ linker mezi opakování III a IV, v „voltage-sensing“ segmentu S4 opakování IV, nebo na vnitřní membráně, kde by mohli narušit dokovací stránky pro inaktivační branky. Klinický fenotyp se liší podle specifické aminokyselinové substituce a, zatímco některé se překrývají, může dojít mezi hyperkalemic PP, paramyotonia congenita (PC), a draslík-zhoršení myotonias (PAM), 3 fenotypy jsou obecně odlišné (jak je popsáno níže). Téměř všechny mutantní kanály narušily rychlou inaktivaci sodíkového proudu. Většina pacientů je citlivá na systémový draslík nebo na chladnou teplotu.
Dvě populace programy existují, mutantní a wild-type; poruchou rychle-inaktivace výsledky v prodloužené depolarizace mutant svalových vláken membrány a může vysvětlit 2 kardinální symptomy těchto poruch, myotonie a slabost. V hyperkalemic PP, a získat funkce se vyskytuje v mutantní kanál vtokové, což vede ke zvýšení sodíku v současnosti příliš depolarizujících postižených svalů. Mírné depolarizace (5-10 mV) myofiber membrány, což může být způsobeno tím, že zvýšená extracelulární koncentrace draslíku, výsledky v mutantní kanály jsou udržovány v noninactivated režimu. Perzistentní proud sodíku dovnitř způsobuje opakované vypalování sodíkových kanálů divokého typu, což je vnímáno jako tuhost (tj.
pokud je přítomna závažnější depolarizace (20-30 mV), normální i abnormální kanály jsou fixovány ve stavu inaktivace, což způsobuje slabost nebo paralýzu. Jemné rozdíly v závažnosti depolarizace membrány tedy mohou znamenat rozdíl mezi myotonií a paralýzou. Teplotní citlivost je charakteristickým znakem PC. Chlad zhoršuje myotonii a vyvolává slabost. S tímto stavem je spojeno několik mutací, z toho 3 na stejném místě (1448) v segmentu S4. Tyto mutace nahrazují arginin jinými aminokyselinami a neutralizují tento vysoce konzervovaný kladný náboj S4. Mutace těchto zbytků jsou nejčastější příčinou PC. Některé z možných mechanismů odpovědných za citlivost na teplotu zahrnují následující:
-
teplota může odlišně ovlivnit konformační změnu v mutantním kanálu.
-
nižší teploty mohou stabilizovat mutantní kanály v abnormálním stavu.
-
Mutace mohou měnit citlivost kanálu na jiné buněčné procesy, jako jsou fosforylace nebo druhé posly.
Většina případů hyperkalemic PP jsou vzhledem k 2 mutace v SCN4A, T704M, a M1592V. Mutace v sodíkovém kanálu, zejména u reziduí 1448 a 1313, jsou zodpovědné za paramyotonia congenita. Malá část případů hypokalémické periodické paralýzy je spojena s mutacemi v kodonech 669 a 672 (HypoPP2). U HypoPP2 mutace sodíkových kanálů zvyšují inaktivaci a vytvářejí čistou ztrátu funkčního defektu.
Normokalemic PP se podobá jak HyperPP (citlivost na draslík), tak HypoPP (trvání útoků) a je způsoben mutacemi SCN4A v hlubším umístění snímače napětí DII v kodonu 675. R675 mutace se liší od HypoPP v tom, že tyto mutace za následek depolarizace-aktivní vtokové pórů generování ω-proud s obrácenou závislost napětí jako tento web je vystavena extracelulární stránek na silnější depolarizace.
kalciového kanálu gen
kalciového kanálu gen (CACNL1A3) je komplex 5 podjednotek (alfa-1, alfa-2, beta, gama a delta). Receptor dihydropyridinu (DHP) kosterního svalu je umístěn primárně v příčné tubulární membráně. Alfa-1 podjednotka má vazebná místa pro léky DHP a vede pomalý proud vápníku typu L. To se také podílí excitace-kontrakce (ES) tažné a působí jako snímač napětí prostřednictvím jeho propojení s ryanodine receptor sarkoplazmatického retikula (tj. uvolňování vápníku kanál). Jakékoli změny membránového potenciálu jsou spojeny s intracelulárním uvolňováním vápníku, což umožňuje vazbu EC. Bodové mutace v alfa-1 podjednotce DHP receptoru/kalciového kanálu způsobují hypokalemickou PP (HypoPP1). Dvě mutace genu CACNA1S, R528H a R1239H, jsou zodpovědné za většinu případů hypokalemická PP.
fyziologický základ onemocnění stále není pochopen, ale je pravděpodobnější kvůli selhání excitace spíše než selhání EC vazby. Depolarizace vyvolaná hypokalémií však může snížit uvolňování vápníku, což přímo nebo nepřímo ovlivňuje řízení napětí kanálu inaktivací sodíkového kanálu. Inzulín a adrenalin mohou působit podobným způsobem. Mutace genu kalciového kanálu mají určité podobnosti s mutacemi SCN4A. Mutace modifikují inaktivaci kanálu, ale ne aktivaci závislou na napětí. Záznamy z kultur myotube od postižených pacientů odhalily 30% snížení vápníkového proudu L-typu citlivého na DHP. Kanály jsou inaktivovány při nízkých membránových potenciálech.
mutace kalciového kanálu způsobují ztrátu funkce projevující se sníženou hustotou proudu a pomalejší inaktivací. Jak tato inaktivace souvisí s útoky vyvolanými hypokalémií, není pochopeno. Alespoň u mutace R528H dochází k možné sekundární kanálopatii, která je spojena se snížením draselného proudu citlivého na ATP ze změněné homeostázy vápníku. Spodní proudy spojené s CACNL1A3 mutace může mírně měnit intracelulární homeostázy vápníku, které by mohly ovlivnit vlastnosti a vyjádření K+ kanály, zejména KATP (ATP-senzitivním kaliovým kanálem), patřící do aktivního usměrňovače třídy kanálů. Inzulín také působí v HypoPP snížením tohoto proudu usměrňovače k+ dovnitř.
ztráta nabití snímače napětí představuje většinu případů HypoPP. Sodíkové a vápenaté kanály mají homologní podjednotky alfa tvořící póry. Bodové mutace v CACNL1A3 a SCN4A ovlivňují Argentinské zbytky v snímačích napětí S4 těchto kanálů. Mutace argininu v segmentech S4 jsou zodpovědné za 90% případů HypoPP.
ztráta nabití snímače napětí představuje většinu případů HypoPP. Sodíkové a vápenaté kanály mají homologní podjednotky α tvořící póry. Téměř všechny mutace v Cav1.1 (HypoPP-1) a Nav1.4 (HypoPP-2) neutralizují kladně nabité aminokyseliny v jednom z nejvzdálenějších arginines nebo lysines napětí senzorů. Nav1.4 mutace se nejčastěji nacházejí v napěťových senzorech opakování I, II a III, což způsobuje únik kationtů.
Nahrazení nejvzdálenějších arginin s menší aminokyseliny jako je glycin otevře vodivé cesty na hyperpolarized potenciál, což vede k vnitřní kation proud (kation úniku nebo ω aktuální odlišit od (ω-a) prostřednictvím ion–vedení pórů, je hyperpolarizace-aktivován aktuální monovalentních kationtů přes S4 vtokové pórů, působí proti nápravu K+ proudy) depolarizací nebo destabilizovat klidový potenciál.
segment S4 se během depolarizace pohybuje směrem ven a uzavírá vodivou dráhu. Svalová vlákna se závažnými mutacemi snímače napětí jsou depolarizována nejen během hypokalémie, ale také při hladinách draslíku v normálním rozmezí, což vysvětluje interiktální a trvalou slabost. Těžká myopatie s tukovou náhradou svalové tkáně se běžně vyskytuje u pacientů s Cav1. 1 R1239H (mutace DIV).
glukokortikosteroidy způsobují HypoPP stimulací Na+ K + ATPázy zprostředkované inzulínem a amylinem.
Gen draselného kanálu
vnitřní rektifikace je důležitou vlastností kanálů Kir. Rektifikace zahrnuje napěťově závislé zablokování pórů pórů polyaminy a Mg++ během depolarizace a toto zablokování je odstraněno během potenciálního gradientu během hyperpolarizace. Mutace draslíkových kanálů jsou pozorovány u Andersen-Tawilova syndromu a tyreotoxického PP.
triáda dysmorfních rysů, periodické paralýzy a srdečních arytmií charakterizuje Andersen-Tawilův syndrom. Tento syndrom je spojen s mutacemi v genu kcnj2. Gen KCNJ2 kóduje vnitřně rektifikující draslíkový kanál Kir2. 1. Uvádí se, že mutace draslíkových kanálů v KCNE3 způsobují hypokalemickou PP, ale to nebylo doloženo.
mutace v Kir2. 6 způsobují citlivost na tyreotoxický PP. Epizodická slabost vidět v thyrotoxic PP je podobný jako v HypoPP a Andersen-Tawil syndrom. Tato porucha je nejčastější u Asiatů a latinskoamerických mužů. Tyreotoxická PP je genetická porucha odhalená tyreotoxikózou. Kir2. 6 je primárně exprimován v kosterním svalu. Trijodtyronin zvyšuje transkripci Kcnj18, což může vést ke zvýšené expresi Kir2. 6. PKC je aktivován během tyreotoxikózy kvůli zvýšenému obratu PIP2 a Kir kanály přímo interagují s PIP2 během normálního vstupu. U Andersen-Tawilova syndromu dochází ke snížení afinity PIP2. U tyreotoxického PP žádná z mutací nemění rektifikaci Kir2. 6.