Aicardi-Goutières-Syndrom: Phänotypisches und genetisches Spektrum in einer Reihe von drei Fällen / Anales de Pediatría
Das Aicardi-Goutières-Syndrom (AGS) ist eine seltene Erbkrankheit, deren genaue Prävalenz unbekannt ist. Es wurde erstmals 1984 von Jean Aicardi und Francoise Goutières als progressive Enzephalopathie mit Beginn in den ersten Lebensmonaten beschrieben, die durch Lymphozytose der Liquor cerebrospinalis (CSF) und Verkalkungen in den Basalganglien gekennzeichnet ist.1 Es äußert sich in Reizbarkeit, psychomotorischer Retardierung, Spastik, Dystonie, epileptischen Anfällen, wiederkehrenden Episoden von aseptischem Fieber und Mikrozephalie. Die Mortalität ist während der enzephalopathischen Phase höher, und obwohl sich die Krankheit typischerweise danach stabilisiert, verursacht sie schwere neurologische Folgen. Andere charakteristische Merkmale, die während ihres Verlaufs auftreten können, sind Frostbeulen, Augensymptome (hauptsächlich Glaukom), Herzbeteiligung oder Autoimmunerkrankungen.2 Typ-I-Interferone spielen eine entscheidende Rolle bei der Pathogenese von AGS, bei denen ihre Expression hochreguliert wird, was zu einer erhöhten Produktion führt.3 Aus diesem Grund ist einer der klassischen Laborbefunde bei diesen Patienten ein erhöhter Interferon alfa-Spiegel im Liquor sowie eine Pleozytose und ebenso erhöhte Neopterin- und Biopterinspiegel. Der potenzielle Nutzen der Beurteilung des Expressionsniveaus von Interferon-stimulierten Genen durch Interferon im peripheren Blut als Marker wird derzeit untersucht, da es Hinweise darauf gibt, dass diese Spiegel über die enzephalopathische Phase hinaus hoch bleiben („Interferonsignatur“).3-5 Ein weiteres wichtiges Merkmal ist die Erkennung von Neuroimaging-Anomalien einschließlich Verkalkungen in den Basalganglien und Veränderungen in der weißen Substanz (Abb. 1). Bis heute kennen wir 7 Gene, deren Mutationen zu einer Hochregulation des Interferonweges führen können: ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1 und IFIH1. Heterozygote Mutationen wurden für die Gene TREX1, ADAR und IFIH1 beschrieben, während die Mutationen, die in allen anderen Genen berichtet wurden, homozygot waren.2 Mutationen im IFIH1-Gen wurden zuletzt nachgewiesen (2014)4 und sind daher die am wenigsten häufigen pathogenen Varianten, während Mutationen in den Genen RNASEH2B und TREX1 den höchsten Anteil an diagnostizierten AGS-Fällen ausmachen.
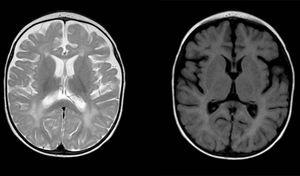
Diffuse und fleckige Signalanomalien in der weißen Substanz in beiden Gehirnhälften, hyperintensiv in T2-gewichteten Bildern. Vergrößerter Subarachnoidalraum mit frontotemporaler Dominanz in beiden Hemisphären, mit Erweiterung der interhemisphärischen Fissur und erhöhter ventrikulärer Größe (ohne erhöhten Druck), kompatibel mit kortikaler und subkortikaler Atrophie.
In den letzten Jahrzehnten hat sich dank der Fortschritte in der Genetik, die den Nachweis dieser spezifischen Mutationen ermöglichen, ein breites phänotypisches Spektrum ergeben, das über die klassische Darstellung des verursachenden Gens hinausgeht. Wir präsentieren die Fälle von 3 Patienten, bei denen in den letzten 8 Jahren AGS diagnostiziert wurde, mit dem Ziel, ihre klinischen Merkmale in Bezug auf den zugrunde liegenden genetischen Defekt zu analysieren (Tabelle 1). Im Allgemeinen stimmten die präsentierenden Merkmale von AGS mit denen überein, die in der jüngsten Fallserie in der Literatur beschrieben wurden: neonatale Präsentation (33%), Mikrozephalie (66%), psychomotorische Retardierung (100%), Spastik (100%), schwere geistige Behinderung (66%) und Verkalkungen auf der Schädel-CT (66%), obwohl nur ein Patient epileptische Anfälle hatte.
Merkmale von Patienten mit Aicardi-Goutières-Syndrom.
| Rechtssache 1 | Rechtssache 2 | Rechtssache 3 | |
|---|---|---|---|
| Genetik | Homozygote Mutation (p.Ala177Thr) im RNASEH2B-Gen | Homozygote Mutation (341G>A) im TREX1-Gen | Heterozygote Mutation (c.992C>G und p.Thr331Arg) im IFIH1-Gen |
| Aktuelles Alter | 3 Jahre | 7 Jahre und 4 Monate | 12 Jahre und 11 Monate |
| Geschlecht | Männlich | Weiblich | Männlich |
| Herkunft | Rumänien | Spanien | Italien |
| AP | – | Woche 36: intrauterine Wachstumsbeschränkung Woche 37: mikrozephalie, Plazentaverkalkung |
Gaumenspalte |
| Klinische Manifestationen | |||
| Alter bei Beginn | 10 Monate | Geburt | 2 Jahre |
| Erstpräsentation | Reizbarkeit Psychomotorische Regression |
Zittern, Hypotonie, schwaches Weinen, Wachstumsstörungen | Verzögerung der motorischen Fähigkeiten |
| Psychomotorische Retardierung | Ja | Ja | Ja |
| Sprache | 2-Silbe words | No | Yes |
| Microcephaly | Yes | Yes | No |
| Chilblains | No | Yes | Yes |
| Epilepsy | No | Yes | No |
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFCS IV |
| Bewegungsstörung | Nein | Ja | Nein |
| Abnormale Augenbewegungen | Nein | Nein | Nein |
| Sehbehinderung | Nein | – | Kurzsichtigkeit |
| Glaukom | Nein | Nein | Nein |
| Hörverlust | – | – | Nein |
| Herzbeteiligung | Nein | Leichte Trikuspidal- und Mitralinsuffizienz | Nein |
| Wiederkehrendes Fieber | Nein | Nein | Nein |
| Geistige Behinderung | Ja | Ja, schwer | Ja, leicht |
| Sonstige | – | – | Singleton-Merten-Syndrom: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
| CSF | |||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – |
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – |
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – |
| INF alfa (IU/mL) | 25 ( | 75 ( | – |
| Blood | |||
| Interferon signature | – | – | Gene overexpression |
| Neuroimaging | |||
| Calcifications on CT | No | Yes. In basalen und periventrikulären Ganglien | Ja. Symmetrische Verkalkungen im tiefen WM der Frontalregion und des Linsenkerns |
| Kopf-MRT | Diffuse und fleckige Veränderungen der WM-Intensität beider Gehirnhälften, hyperintensiv auf T2. Beteiligung von subkortikalem WM (Schonung der U-Fasern) und periventrikulärem WM | Generalisierte WM-Beteiligung mit Vorherrschen von lobärem WM einschließlich der subkortikalen U-Fasern des Frontal-, Temporal- und Okzipitallappens, bilateral und symmetrisch, ohne kortikale Beteiligung | – |
Liquor, Liquor cerebrospinalis; CT, Computertomographie; GMFCS, Klassifizierungssystem der Grobmotorik; INF, Interferon; IUGR, intrauterine Wachstumsrestriktion; MRT, Magnetresonanztomographie; PNP, Polyneuropathie; WM, White mater.
Wie bereits erwähnt, sind homozygote Mutationen im RNASEH2B-Gen die häufigsten Varianten, die AGS verursachen, und ihre phänotypische Expression entspricht normalerweise am meisten der klassischen Präsentation.4 Dies war der Fall bei dem Patienten in unserer Studie, der eine solche Mutation trug und im Alter von 10 Monaten mit Reizbarkeit und psychomotorischer Retardierung sowie mit charakteristischen Neuroimaging- und Liquorbefunden auftrat.
Zwanzig Prozent der Fälle von AGS können eine neonatale Präsentation haben, wobei der Ausbruch der Krankheit in utero auftritt.5 Mutationen in einem der 7 oben genannten Gene können zu diesem Phänotyp führen, aber diese frühe Präsentation ist am häufigsten mit dem TREX-Gen assoziiert.4,5 Die anfängliche Darstellung dieser Form ähnelt der einer TORCH-Infektion mit Hepatosplenomegalie, Hypertransaminasämie, Thrombozytopenie und neurologischen Manifestationen einschließlich extremer Reizbarkeit, Bewegungsstörungen und epileptischen Anfällen.5 Diese Patienten haben einen schwereren Krankheitsverlauf und ein höheres Sterberisiko. Der Patient in unserer Stichprobe, der sich mit einer solchen Variante vorstellte, hatte eine neonatale Präsentation und hat derzeit die schwerste Form der Erkrankung der 3.
Mutationen im ADAR1-Gen und insbesondere im IFIH1-Gen sind mit einem späten Einsetzen der Symptome nach 1 Lebensjahr mit normaler psychomotorischer Entwicklung verbunden.5 In einigen dieser Fälle hat das Syndrom einen gutartigen Verlauf mit relativer Erhaltung der Sprache und motorischen Fähigkeiten. Unser Patient mit einer Mutation im IFIH1-Gen war insofern ein Einzelfall, als er auch das Singleton-Merten-Syndrom hatte, eine seltene Krankheit, die ebenfalls durch eine Mutation im IFIH1-Gen verursacht wurde und durch Zahndysplasie, Aortenverkalkungen und Osteoporose gekennzeichnet ist.6
Unser Ziel ist es, die signifikante phänotypische Variabilität von AGS und seine Assoziation mit spezifischen Mutationen zu unterstreichen, um sowohl die Berücksichtigung dieser Diagnose bei von der klassischen Krankheitsform abweichenden Darstellungen zu fördern als auch zusätzliche Informationen über den Krankheitsverlauf und die Ergebnisse bei diesen Patienten beizutragen.