Bioorganische und organische Chemie
Hydroborierungsoxidationsreaktion
Die cis–Addition von Diboran an eine Alkenbindung bietet eine äußerst nützliche Hydratationsmethode. Diboran kann durch Zugabe von Natriumborhydrid zu Bortrifluoridetherat in Tetrahydrofuran oder Ether bei 0o-5oC erzeugt werden. Diboran ist das Dimer von Boran (BH3) und ist stabile Form dieses Reagens (Scheme1).
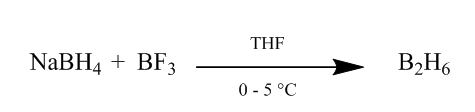
Abbildung 1: Gewinnung von Diboran aus Natriumborhydrid
Die Zugabe von Diboran zum Alken erfolgt extrem schnell und im Allgemeinen addiert sich das Reagenz von der weniger gehinderten der beiden Seiten des π-Systems. Die cis-Addition wurde durch einen Übergangszustand mit vier Zentren rationalisiert. Der aus der Addition von Diboran an ein Alken resultierende Borankomplex wird unter Beibehaltung der Stereochemie durch Behandlung mit basischem Wasserstoffperoxid in einen Alkohol überführt. So führt 1-Methylcyclohexen 1 bei der Hydroborierung-Oxidation zur Bildung von trans-2-methylcyclohexanol 2. Der mechanistische Weg ist in (Schema 2) dargestellt. Die Methode zur Umwandlung von Alken in Alkohol durch Hydroborierung-Oxidation wurde für die Synthese vieler Naturprodukte angewendet. Einige Beispiele sind unten dargestellt.

Abbildung 2 Reaktionsmechanismus für die Bildung zu trans-2-Methylcyclohexanol 2
Synthese von (±) junenol und (±) acalomon
Die Verwendung einer Hydroborierungsoxidationsreaktion wurde von Banerjee und Mitarbeitern während der Synthese3 von Eudesmon-Sesquiterpenen (±) Junenol und (±) Acalomon beobachtet. Um die Synthese dieser Sesquiterpene zu erreichen, wurde das Alken 3 als Ausgangsmaterial ausgewählt und unter Hydroborierung zum Alkohol 4 oxidiert (Schema 3). Das durch Oxidation des Alkohols mit Jones-Reagenz 4 erhaltene Keton 5 wurde mit Diethylcarbonat zur Reaktion gebracht. Das resultierende Produkt wurde mit Methyllithium behandelt, um das Ketol 6 zu erhalten, dessen Umwandlung zum Isopropylketon 7 durch Dehydratisierung bzw. Hydrierung erfolgte. Durch Metallhydridreduktion des Ketons und anschließende Oxidation mit Bleitetraacetat 5 in Cyclohexan erhielt man den cyclischen Ether 8, der durch Oxidation mit Chromsäure und Essigsäure in Ketosäure 9 überführt wurde. Decarboxylierung mit Bleitetraacetat in Benzol und Pyridin gefolgt von Reinigung über 10% AgNO3 imprägniertes Kieselgel ergab (±) Acolamon 10. Reduktion von Acolamon 10 mit Natriumborhydrid in Methanol und anschließende Sublimation des erhaltenen Produktes ergab Junenol 11.
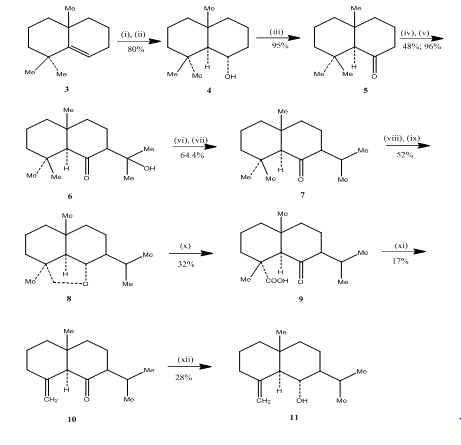
Abbildung 3 Synthese der Eudesmon-Sesquiterpene (±) junenol und (±) -acalomon
Reagenzien: (i) BF3.Et2O, NaBH4, THF, 0-5 ° C; (ii) NaOH (10%), H2O2 (30%); (iii) CrO3 / HMPT; (iv) NaH, CO (OEt) 2, DME; (v) MeLi, Et2O, Reflux, 2h; (vi) HCl (konz.), MeOH; (vii) H2, PtO2, MeOH; (viii) Na, EtOH, Rückfluss; (ix) Pb(OAc)4, C6H12; (x) CrO3, AcOH; (xi) Pb(OAc)4, C6H6, Py, Rückfluss; (xii) NaBH4, EtOH.
Synthese von Pisiferinsäure
Bei der Synthese von Pisiferinsäure, einem tricyclischen Diterpen, das antibakterielle Aktivitäten gegen alle getesteten grampositiven Bakterien zeigt, wurde die Verwendung von Hydroborierungsoxidation aufgezeichnet.7 ist der Syntheseweg in Schema 4 dargestellt. Die Hydroborierung-Oxidation des Alkens 13, hergestellt aus dem bekannten 8 Ketoalkohol 12, wurde mit Jones-Reagenz 4 oxidiert bzw. mit Metallhydrid zu Alkohol 14 reduziert. Oxidation mit Bleitetraacetat in Benzol mit 250W Wolframlampe ergab den cyclischen Ether 15. Die Spaltung des cyclischen Ethers mit Zink, Zinkiodid und Essigsäure lieferte Pisiferol 16. Die Umwandlung des Pisiferols in den Ester 17 erfolgte in sechs Schritten:
- Methylierung mit Dimethylsulfat
- Oxidation mit Jones-Reagenz
- Veresterung mit Diazomethan
- Reduktion mit Natriumborhydrid
- Tosylierung
- Detosylierung
Der Ester l7 wurde durch Erhitzen mit Aluminiumbromid und Ethanthiol in Pisiferinsäure 18 überführt.
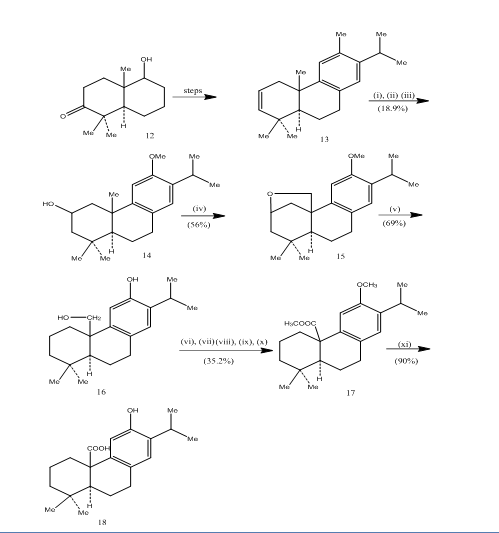
Abbildung 4 Synthese von Pisifersäure 18
Reagenzien: (i) BF3.Et2O, NaBH4; (ii) NaOH (10%), H2O2 (30%), H2SO4-HCrO4; (iii) LiAlH4, THF; (iv) Pb (OAc) 4, CaCO3, C6H6, 250w Wolframlampe; (v) Zn, ZnI, MeCOOH; (vi) MeSO4, Me2CO; (vii) H2SO4-HCrO4; (viii) CH2N2, Et2O; (ix) NaBH4, MeOH; (x) TsCl, Py; (xi) NaI, Zn Staub, DMF; (xii) AlBr3, (CH2SH)2.
Die Hydroboration-Oxidationsreaktion ist für die Synthese von (±) eudes angewendet worden-4(14),7(11)- dien-8-on, 9 Taxodion, 10 Norditerpenalkohole11 und viele andere Terpene.12 Diese Beispiele zeigen deutlich die Verwendung von Bortifluoridetherat bei der Umwandlung der Alkene in Alkohole und anschließend deren Umwandlung in die Terpenoidverbindungen.
Spaltung von Epoxiden
Die Epoxide können durch mehrere Reagenzien gespalten werden. Das Lewis-Säure-Bortrifluorid-Etherat wurde auch zur Spaltung von Epoxiden verwendet, und in vielen Fällen ordnet sich das resultierende Produkt zu Keton um. Die Spaltung von Epoxiden geht ebenfalls mit einer Cyclisierung einher. In dieser Übersicht wurde die Spaltung einiger Epoxide mit Bortrifluoridetherat und die Verwendung der resultierenden Produkte bei der Synthese von Naturstoffen diskutiert.
Synthese von 6-Methoxy-2-tetralon
Die Spaltung von Epoxid mit Bortrifluoridetherat wurde für die Synthese von 6-Methoxy-2-tetralon 20 (Schema 5), einem wichtigen ausgewählten Ausgangsmaterial für die Synthese vieler organischer Verbindungen, verwendet13. Durch Epoxidierung des Alkens 13 19 und anschließende Behandlung des Rohproduktes in Dichlormethan mit Bortrifluoridetherat wurde das Tetralon 20 in 36%iger Ausbeute erhalten. Beim Versuch der Spaltung mit Schwefelsäure verbesserte sich die Ausbeute des Teralons 20 (39%) zusammen mit der Bildung anderer Nebenprodukte, so daß die chromatographische Reinigung sehr aufwendig war.
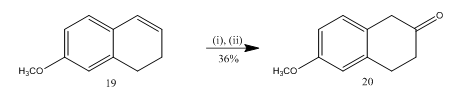
Abbildung 5 Synthese von 6-Methoxy-2-tetralone 20
Reagenzien: (i) MCPBA, CH2Cl2; (ii) BF3OEt2
Synthese von Cupran
Die Umlagerung von Epoxiden durch Bortrifluoridetherat erwies sich während der Synthese14 von Sesquiterpen-Cupran als sehr nützlich. Der Syntheseweg ist in Schema 6 beschrieben. 6,6-dimethyl-1-p-tolylcyclohexen 21 lieferte bei Epoxidierung das Epoxid 22 in guter Ausbeute, das bei Behandlung mit bortrifluoridetherat in Benzol den Aldehyd 23 in geringer Ausbeute lieferte. Das Semicarbazon des Aldehyds wurde mit Kaliumhydroxid erhitzt, um das Sesquiterpen-Cupran 24 in akzeptabler Ausbeute zu liefern. Die Synthese ist aufgrund ihrer Kürze in Schritten attraktiv. Die Bedingungen für die Umlagerung des Epoxids 22 sind kritisch, da es zur weiteren Umlagerung zum Keton 25 neigt.
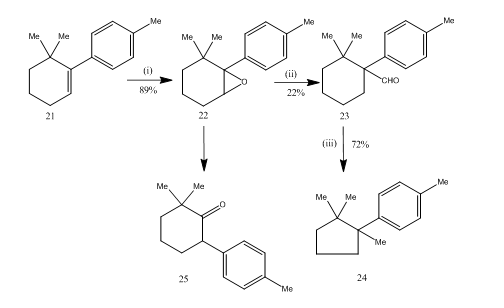
Abbildung 6 Synthese von cuprane
Reagenzien: (i) PhCO3H, CHCl3; (ii) C6H6, BF3Et2O; (iii) NH2NHCONH2, KOH
Synthese von (±) Karahana-Ether
Bortrifluoridetherat wurde auch für die Spaltung von Epoxid während der Synthese15 von Karahana-Ether, einem flüchtigen Monoterpen, das aus japanischem Hopfen isoliert wurde16, verwendet. Der Syntheseweg ist in Schema 7 beschrieben. Das aus dem Dien 26 erhaltene Epoxid 27 wurde bei der Behandlung mit Bortrifluoridetherat cyclisiert und erhielt das Produkt 28. Die Cyclisierung erfolgte wahrscheinlich durch das Zwischenprodukt 27(i). Die Metallhydridreduktion lieferte Diol, das bei der Tosylierung Karahanaether 29 ergab. Die Ausbeute ist nicht spezifiziert. Die Spaltung von Epoxiden wurde für die Synthese vieler Terpene wie Rosenolacton, 17 Cyperolon, 18 Maritimol genutzt.19
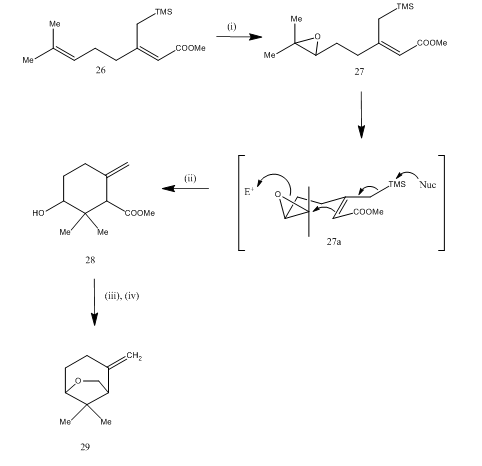
Abbildung 7 Synthese von (±) Karahana Ether
Reagenzien: (i) MCPBA; (ii) BF3Et2O; (iii) LiAlH4; (iv) TsCl, Py
Veresterung
Die Veresterung ist eine häufig verwendete Reaktion zur Synthese vieler organischer Verbindungen. Bortrifluoridetheratalkohol ist ein sehr geeignetes Reagenz für die Veresterung vieler p-Aminobenzoesäuren, aromatischer, heterocyclischer und ungesättigter Säuren.20 In einigen Veresterungsreaktionen lieferte die Verwendung dieses Reagens eine überlegene Ausbeute im Vergleich zu anderen Reagenzien. Einige Beispiele sind in Schema 8 angegeben. Die Säuren 30-32 wurden bei Behandlung mit Bortrifluorid-Etherat-Alkohol-Reagenz in hoher Ausbeute in die Ester 33-35 überführt. Marshall und mitarbeiter21 verwendeten dasselbe Reagenz für die Veresterung von Carbonsäuren. Dymicky22 stellte mehrere Formate in hoher Ausbeute aus Ameisensäure und Alkohol in Gegenwart einer katalytischen Menge an Bortrifluorid-Methanol-Komplex her. Die anderen Katalysatoren z.B. Schwefelsäure, p-Toluolsulfonsäure war nicht so effizient wie Bortrifluorid-Methanol-Komplex.
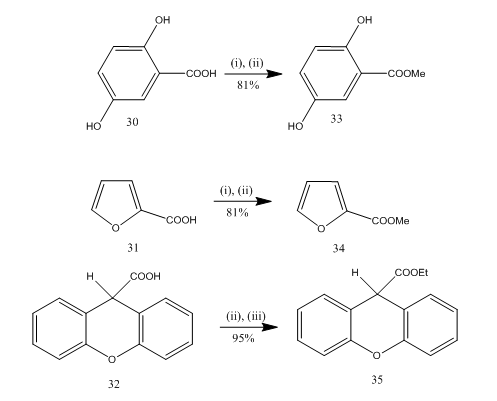
Abbildung 8 Veresterung von Säuren 32-35
Reagenzien: (i) MeOH; (ii) BF3.Et2O; (iii) EtOH
Jackson und mitarbeiter23 haben eine effiziente Methode zur Umwandlung von Alkoholen 37-39 und Säuren 40-42 direkt in die entsprechenden t-Butylderivate in guter Ausbeute unter Verwendung von t-Butyltrichloracetimidat 36 in Gegenwart einer katalytischen Menge an Bortrifluoridetherat entwickelt, wie in Schema 9 gezeigt. Diese Methode funktioniert mit den säureempfindlichen Gruppen besser als die traditionellen Methoden unter Verwendung von Isobuten. Eine weniger gehinderte Hydroxylgruppe eines Diols kann geschützt werden und ist auch für Arbeiten in kleinem Maßstab geeignet (Vermeidung der Verwendung von gasförmigem Isobuten). Das t-Butyl-2,2,2-trichloracetimidat 36 läßt sich leicht durch Zugabe von t-butanol zu Trichloracetonitril herstellen. Die meisten Versuche wurden in Gegenwart eines Gemisches aus Dichlormethan und Cyclohexan durchgeführt. Essigsäureanhydrid in Gegenwart von Bortrifluoridetherat wurde für die Acetylierung der Hydroxylgruppe verwendet.24

Abbildung 9 Umwandlung von Alkoholen und Säuren aus T-Butylderivaten.
Reagenzien: 36, (i) BF3.Et2O, (ii) CH2Cl2, C6H12
Cyclisierung
Das Bortrifluoridetherat hat eine wichtige Rolle bei der Cyclisierung vieler Carbonsäuren, Allene usw. gespielt. Die folgenden Beispiele sollen die Rolle von Bortrifluoridetherat als Cyclisierungsmittel verdeutlichen. Das Säurechlorid 49 und das Alken 50 wurden zu Divinylketon kondensiert25 51, das einer Nazarov-Cyclisierung unterlag26,27 und cyclischem Keton 52, das in das Sesquiterpentrichodien 53 umgewandelt wurde (Schema 10).
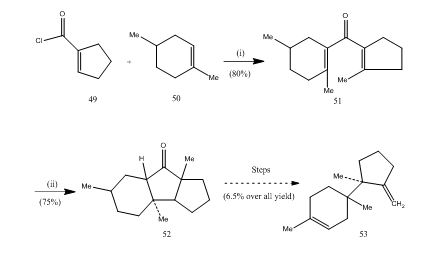
Abbildung 10 Synthese von Sesquiterpentrichodien 53
Reagenzien: (i) SnCl4, NaOMe; (ii) BF3Et2O
Mehrere Allenylarylketone durchlaufen eine Cyclisierung mit Bortrifluoridetherat, wobei Methylenbenzocyclopentenon über eine neue 5-Endomode-Cyclisierung erhalten wird.28 Die Ketone 54-56 ergaben jeweils Benzocyclopentenone 57-59 (Schema 11). Wahrscheinlich erfolgte die Umwandlung wie bei der Cyclisierung von Allenylarylketon 54 zu 57 gezeigt. Es kann beobachtet werden, dass die Anwesenheit von Substituentengruppen im aromatischen Ring die Ausbeute des cyclisierten Produkts bestimmt. Kos und Loewenthal28 berichteten über die Cyclisierung der Säure 60 mit Bortrifluoridetherat zum Keton 61, das Gibberon 62 umgesetzt wurde (Schema 12) in drei Schritten:
- Ketalisierung
- Huang-Minlon-Reduktion und
- Säurehydrolyse. Die oben genannten Beispiele zeigen die Verwendung von Bortrifluoridetherat bei der Cyclisierung organischer Verbindungen
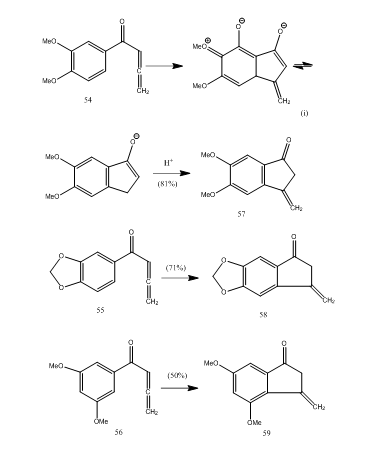
Abbildung 11 Synthese von Cyclopentenonen

Abbildung 12 Synthese zu Gibberon
Reagenzien: (i) BF3.Et2O; (ii) (a) C2H6O2; (b) GRAD, N2H4, KOH, 190-200 ° C; (c) H+