Fallbericht Gynäkologie und Geburtshilfe
Schlüsselwörter
Tetra-Amelia; Fehlbildung; Genotyp; Phänotyp.
Einleitung
Extremitätenanomalien stellen eine wichtige Gruppe angeborener Pathologien dar, die durch Hypoplasie oder vollständige Aplasie eines oder mehrerer Extremitätenknochen gekennzeichnet sind. Extremitätenanomalien aller Art treten bei etwa 1 von 1.300 bis 2.000 Geburten auf. Diese Extremitätenanomalien können isoliert oder mit anderen Anomalien assoziiert sein . Tetraamelia-Syndrom ist selten und Grauzonen bleiben.
Wir berichten über zwei Fälle von Tetra-Amelia in einer Mutterschaft der Stufe II in Dakar (Senegal) mit Tetraamelia-1 (Chromosom 17q21), Tetraamelia-2 (Chromosom 8q23) und Robert-Syndrom (Chromosom 8p21). Dies zeigt die Schwierigkeit bei der Korrelation von Phänotyp und beteiligten Genen.
Fallberichte
Fall 1
Frau AD war eine 44-jährige Mutter, die in der 36. Schwangerschaftswoche mit schwerer Präeklampsie und fetalen Anomalien an unsere Abteilung überwiesen wurde. Sie war fünf Jahre alt und hatte keine fetalen Anomalien in der Vorgeschichte. Sie rauchte jetzt nicht und hatte noch nie Alkohol geraucht oder getrunken. Sie war keinem Passivrauchen ausgesetzt. Sie war in einer blutsverwandten Ehe dritten Grades für alle ihre Kinder. Frau AD hatte negativ auf Hepatitis B, HIV und Syphilis getestet. Sie war vor dem Rötelnvirus geschützt und hatte zuvor keine Exposition gegenüber Toxoplasma gondii. Die Ultraschallüberwachung, die spät nach 33 Wochen und 35 Wochen der Schwangerschaft durchgeführt wurde, ergab Oligoamniose und Hydrozephalus sowie Agenese der Gliedmaßen. Verschreibungen während der Schwangerschaft umfassten die Verabreichung von Eisen und Folsäure sowie die Verabreichung von Sulfadoxinpyrimethamin. Letzteres wurde nach 18 Wochen und dann nach 26 Wochen im Rahmen der Anti-Malaria-Prophylaxe für schwangere Frauen verschrieben. Die symphyseal-fundale Höhe betrug 28 cm. Aufgrund schwerer Merkmale der Präeklampsie; Sie wurde sofort ins Krankenhaus eingeliefert und in einer Arbeits- und Entbindungseinheit beobachtet. Sie erhielt dann zunächst IV Magnesiumsulfat, um Eklampsie und blutdrucksenkende Medikamente zu verhindern, um den systolischen Blutdruck unter 160 mmHg und den diastolischen Blutdruck unter 105 mmHg zu halten.
Die Entscheidung der sofortigen Kaiserschnitt wurde getroffen. Ein 2.150 Gramm Lebendgeborener wurde extrahiert, der anschließend innerhalb von 10 Minuten starb. Vor dem Tod zeigte der Körper kriechende Bewegungen. Es wurden mehrere äußere Anomalien identifiziert (Abbildung 1), einschließlich der vollständigen Agenese aller vier Gliedmaßen, Hydrozephalus mit einem Kopfumfang von 39 cm. Im Gesicht gab es Hypertelorismus mit einem Kolobom der Augenlider, leichten Exophthalmus und Aniridien. Der Mund war wie ein umgekehrtes V ohne klare Abgrenzung der Lippen und die Nase war rudimentär. Das Neugeborene war frei von Integumenten (Haare und Augenbrauen). Die Ohren waren auf Skizzen reduziert, die wie Schlitze aussahen. Der Hals war kurz. Der Stamm wurde auf eine 26 cm lange konische Struktur mit einer Nabelschnur am unteren Ende reduziert. Die Brust war schmal. Knapp unterhalb des Nabels befand sich am Boden des Schwanzpols der nach hinten erweiterte Rumpf, eine Mittellinie mit Rezessionen und eine Knospung, die einem Phallus unbestimmten Typs entsprechen kann. Agenesis des Beckens, Genitalien und anal Imperforation wurden festgestellt. Fetale Pathologie wurde nicht durchgeführt. Der Tod innerhalb von 10 Minuten nach der Entbindung und das konische Erscheinungsbild der Brust können jedoch auf Lungenanomalien hindeuten.
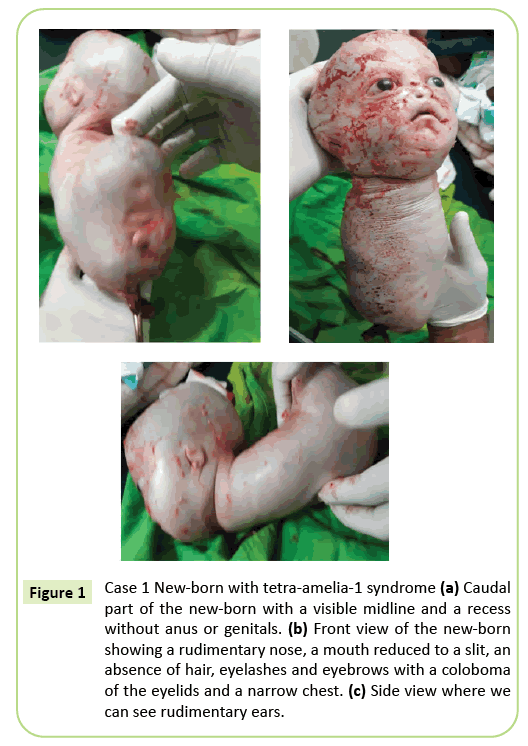
Abbildung 1: Fall 1 Neugeborenes mit Tetra-Amelia-1-Syndrom (a) Kaudaler Teil des Neugeborenen mit sichtbarer Mittellinie und Aussparung ohne Anus oder Genitalien. (b) Vorderansicht des Neugeborenen mit einer rudimentären Nase, einem zu einem Schlitz reduzierten Mund, einem Fehlen von Haaren, Wimpern und Augenbrauen mit einem Kolobom der Augenlider und einer schmalen Brust. (c) Seitenansicht, wo wir rudimentäre Ohren sehen können.
Fall 2
Der zweite Fall war eine 22-jährige Primigravida, die nach 37 Schwangerschaftswochen zur Ultraschalluntersuchung an unsere Einrichtung überwiesen wurde. Sie war nicht in einer blutsverwandten Ehe. Sie hatte negativ auf Hepatitis B, HIV und Syphilis getestet. Sie wurde nicht auf Toxoplasmose und Röteln getestet. Während ihrer Schwangerschaft wurde keine Ultraschallüberwachung durchgeführt. Die klinische Untersuchung stimmte mit einer fetalen Wachstumsverzögerung überein (Fundalhöhe: 26 cm). Ultraschallbefunde zeigten eine Verzerrung des Humerus von 23,9 mm, was einer Schwangerschaft von 17 Wochen entspricht. Es gab eine Agenese des Femurs. Die Beckenflügel waren im Ultraschall sichtbar. Es wurden keine Lungen- oder Herzanomalien festgestellt. Die Lieferung wurde eingeleitet. Das Neugeborene hatte einen weiblichen Phänotyp mit einem Apgar-Score von 9 in der 5. Minute. Die Morphologie des Kopfes und des Rumpfes war ohne Besonderheit. Die oberen Gliedmaßen wurden auf zwei 3 cm lange Stümpfe reduziert. Es wurde eine vollständige Agenese der 2 unteren Gliedmaßen festgestellt. Es war eine symmetrische Anomalie (Abbildung 2).
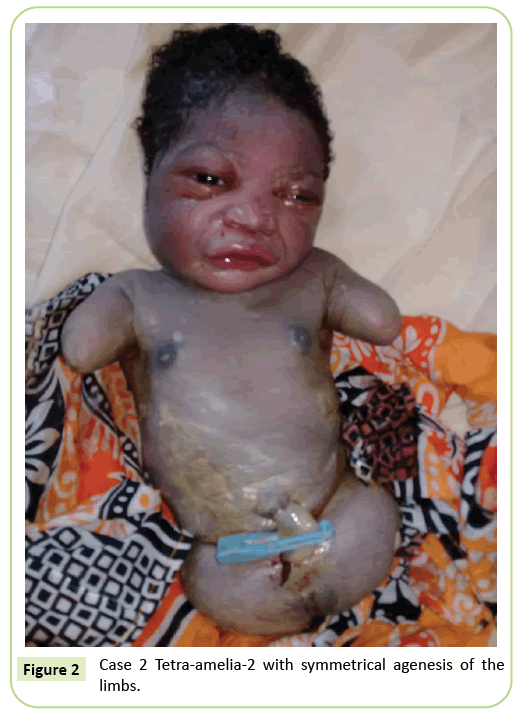
Abbildung 2: Fall 2 Tetra-amelia-2 mit symmetrischer Agenese der Gliedmaßen.
Diskussion
Bermejo-Sanchez et al. 2011 wurde die Epidemiologie der angeborenen Amelia anhand von Daten beschrieben, die zwischen 1968 und 2006 aus 20 Überwachungsprogrammen für angeborene Anomalien auf allen Kontinenten außer Afrika gesammelt wurden. Insgesamt wurden 326 Fälle von Amelia unter 23.110.591 Lebendgeburten, Totgeburten und Abtreibungen identifiziert. Die Prävalenz betrug 1,41 / 100.000 .
Tetra-Amelia bezeichnet das völlige Fehlen der Gliedmaßen und tritt seltener auf. Unseres Wissens ist Tetra-amelia-1 in 7 Familien beschrieben. Es scheint einer autosomal-rezessiven Vererbung zu folgen. In allen Familien war Tetra-Amelia-1 mit schweren Missbildungen der anderen Körperteile einschließlich Gesicht und Kopf, Anomalien des Nervensystems, des Skeletts und der Genitalien verbunden. Die Lunge war in vielen Fällen unterentwickelt, was das Atmen erschwert oder unmöglich macht . Zimmer et al. berichtet wurde 1985 eine stark Inzuchtfamilie, in der 6 Säuglinge Tetra-Amelia-1 und Hydrocephalus hatten. Sie beschrieben bei einem der Föten ein völliges Fehlen von Beckenknochen, Lippen- und Gaumenspalte, Arrhinia und Aplasie der Ohren. Eine bilaterale linke Lunge, ein persistierender arterieller Kanal, eine anale Imperforation wurden ebenfalls festgestellt. Fetale Tests beseitigten die Diagnose des Robert-Syndroms . Andere in der Literatur gefundene Fälle umfassen die von Kosaki et al., 1996, mit einem Fötus des Karyotyps 46, XX mit Tetraphokomelie und schwerer Lungenhypoplasie zusätzlich zu Gesichts- und Kopfanomalien . Rosenak et al. beschrieb einen Fall von Tetra-Amelia mit schwerer Lungenhypoplasie bei zwei Föten eines nicht blutsverwandten Paares. Fetale Tests schlossen die Diagnose des Robert-Syndroms aus . Zwei weitere Fälle wurden von Zlotogora et al. im Jahr 1993. Beide Patienten starben kurz nach der Geburt und die Autoren schlugen die Existenz einer Lungenhypoplasie vor. Niemann et al. berichtete eine blutsverwandte türkische Familie, in der 4 der 8 Brüder an Tetra-Amelia litten. Zusätzlich zum Fehlen der 4 Gliedmaßen ergaben die fetalen Untersuchungen von 3 Föten mehrere Anomalien: Lippen- und / oder Gaumenspalte, Laparoschisis, Lungenanomalien, Hypoplasie des Beckens, Atresie der Choanas, Vagina und Analimperforation . Schließlich, im Jahr 2005, Krahn et al. beschrieben 2 Brüder von Inzuchteltern, die an Tetraamelie und schwerer Lungenhypoplasie leiden. Die Schlüsselbeine und Schulterblätter waren beim zweiten Fötus normal. Der Karyotyp war normal .
Das Tetra-Amelia-1-Syndrom oder TETAMS1 wird durch eine homozygote Mutation im WNT3-Gen auf Chromosom 17q21 mit autosomal-rezessiver Vererbung verursacht. Das Tetraamelia-2-Syndrom (TETAMS2) ist durch rudimentäre Gliedmaßen oder ein vollständiges Fehlen der Gliedmaßen gekennzeichnet, im Allgemeinen symmetrisch sowie in einigen Fällen bilateral Agenese der Lunge. Sind auch übliche Anomalien des Lungengefäßsystems und Dysmorphien einschließlich bilateraler Lippen- und Gaumenspalte, Ankyloglossie, Unterkieferhypoplasie, Mikroretrognathie und labioscrotal Aplasie .
Szenker-Ravi, der 4 Familien von Tetra-Amelia mit Agenese oder pulmonaler Hypoplasie untersuchte, stellte eine phänotypische Heterogenität mit Extremitätenanomalien unterschiedlicher Schwere fest . Die Exomsequenzierung in diesen 4 Familien hat es ermöglicht, homozygote Mutationen im RSPO2-Gen zu identifizieren . Das Tetraamelia-2-Syndrom wird durch eine homozygote Mutation im RSPO2-Gen (610575) auf Chromosom 8q23 verursacht .
Der Phänotyp des ersten in diesem Artikel beschriebenen Falles entspricht einem Tetra-Amelia-1-Syndrom, das insbesondere auf das Vorhandensein von Hydrozephalus, Anomalien der Genitalien und einer rudimentären Nase zurückzuführen ist. Die schmale Brust und der frühe Tod vor der 10. Dieser Fall hebt die phänotypische Heterogenität mit einem Augenlidkolobom, Hypertelorismus, Exophthalmus und seltenen Anhängseln hervor.
Wir betrachten den zweiten Fall in unserer Studie als Tetramelia-2-Syndrom unter Berücksichtigung der symmetrischen Tetra-Amelia mit dem Vorhandensein von Stümpfen der oberen Extremitäten. Die Diagnose von Tetra-Amelia sollte früh während der Ultraschallüberwachung erfolgen. Daher sollte das Bewusstsein für die Bedeutung der Ultraschallüberwachung und der Verwendung von 3D / 4D zur Verbesserung der Screening-Ergebnisse geschärft werden. Die Diagnose einer Beckenmasse im Ultraschall gepaart mit Amelia sollte den Verdacht auf ein Spleno-Gonaden-Fusion-Gliedmaßendefektsyndrom erwecken.
Darüber hinaus sind die fetale Untersuchung und fetale Tests mit den sich entwickelnden Technologien des chromosomalen Microarrays und der Exom- und Genomsequenzierung in unseren Einstellungen zu fördern. Eine bessere Charakterisierung der Fälle ermöglicht es, Paare zu beraten und diese klinischen Anomalien besser zu kennen.
Fazit
Das Tetra-Amelia-Syndrom ist selten und es bleiben immer noch Grauzonen. Diese beiden Fälle veranschaulichen im Vergleich zu dem, was bereits in der Literatur beschrieben ist, die phänotypische Heterogenität von Tetraamelia. Angesichts der seltenen Inzidenz dieser Anomalien wäre es wichtig, ein internationales Anomalieregister zu erstellen, um Fälle zu melden, und eine Probenbank für erweiterte genetische Studien an die Eltern einzurichten.
- Wilcox WR, Coulter CP, Schmitz ML (2015) Angeborene Gliedmaßenmangelerkrankungen. Clin Perinatol 42: 281-300.
- Bermejo SE, Cuevas L, Amar E (2011) Amelia: Eine multizentrische deskriptive epidemiologische Studie in einem großen Datensatz des International Clearinghouse for Birth Defects Surveillance and Research und Überblick über die Literatur. Bin J Mit Genet C Semin Mit Genet 157: 288-304.
- Zlotogora JSM, Shabany YO, Jarallah RY (1993) Syndrom der Tetraamelie mit pulmonaler Hypoplasie. Bin J Med Genet 47: 570-571.
- Zimmer EZ (1985) Tetra-Amelia mit multiplen Missbildungen bei sechs männlichen Föten in einer Verwandtschaft. Europ. JOH 144,412-414.
- Rosenak D (1991) Rezidivierende Tetraamelien und pulmonale Hypoplasie mit multiplen Missbildungen bei Sibs. Am J Med Genet 38: 25-28.
- Gershoni BR (1990) Roberts-Syndrom oder „X-verknüpfte Amelia“ ? . Bin J Med Genet 37: 569-572.
- Kosaki K, Jones MC, Stayboldt C (1996) Zimmer Phocomelia: Abgrenzung durch Hauptkoordinatenanalyse. Bin J Med Genet 66: 55-59.
- Niemann S(2004) Homozygote WNT3-Mutation verursacht Tetra-Amelia in einer großen blutsverwandten Familie. Bin J Hum Genet 74: 558-563.
- Krahn M (2005)Tetra-Amelia- und Lungenaplasie-Syndrom: Bericht über eine neue Familie und Ausschluss von Kandidatengenen. Clin Genet 68: 558-560.
- Szener-RE, Altunoglu U (2018) RSPO2 Die Hemmung von RNF43 und ZNRF3 regelt die Entwicklung von Gliedmaßen unabhängig von LGR4 / 5 /6. Natur 557: 564-569.