Periodische Lähmungen
Eine klinisch nützliche Klassifikation der primären periodischen Lähmungen, die in Tabelle 1 gezeigt wird, umfasst hypokaliämische, hyperkalämische und paramyotonische Formen.
Tabelle 1. Primäre periodische Lähmung (modifiziert von Jurkat-Rott und Lehmann-Horn ) (Tabelle in neuem Fenster öffnen)
|
Krankheit |
Gen |
Proteine |
Vererbung |
Mutation |
|
HyperPP |
SCN4A |
11.4 |
Dominant |
Fein |
|
NormoPP |
Fein (ω-Pore) |
|||
|
Paramyotoniacongenita |
Fein |
|||
|
HypoPP Typ II |
Fein (ω-Pore) |
|||
|
HypoPP Um |
CACNA1S |
11.1 |
Dominant |
Verstärkung (ω-)) |
|
Thyreotoxikose |
Modell: KCNJ18 |
Kir2.18 |
Dominant |
Verlust |
|
Andersen-Tawil-Syndrom |
Modell: KCNJ2 |
Kir2.1 |
Dominant |
Verlust |
Die physiologische Grundlage der schlaffen Schwäche ist Inerregbarkeit der Muskelmembran (dh Sarkolemma). Die Veränderung des Serumkaliumspiegels ist nicht der Hauptdefekt des primären PP; Der veränderte Kaliumstoffwechsel ist ein Ergebnis des PP. Bei primärer und thyreotoxischer PP tritt eine schlaffe Lähmung mit relativ geringen Veränderungen des Serumkaliumspiegels auf, während bei sekundärer PP die Serumkaliumspiegel deutlich abnormal sind.
Kein einziger Mechanismus ist für diese Gruppe von Störungen verantwortlich. Sie sind also heterogen, weisen jedoch einige gemeinsame Merkmale auf. Die Schwäche ist normalerweise verallgemeinert, kann aber lokalisiert sein. Schädelmuskulatur und Atemmuskulatur werden in der Regel geschont. Dehnungsreflexe sind während der Angriffe entweder nicht vorhanden oder vermindert. Die Muskelfasern sind während der Angriffe elektrisch nicht erregbar. Die Muskelkraft ist zwischen den Anfällen normal, aber nach einigen Jahren entwickelt sich bei bestimmten PP-Typen (insbesondere primärem PP) ein gewisses Maß an fester Schwäche. Alle Formen der primären PP (außer Becker Myotonia congenita ) sind entweder autosomal-dominant vererbt oder sporadisch (höchstwahrscheinlich aufgrund von Punktmutationen).
Spannungsempfindliche Ionenkanäle regulieren eng die Erzeugung von Aktionspotentialen (kurze und reversible Veränderungen der Spannung von Zellmembranen). Dies sind selektiv und variabel durchlässige Ionenkanäle. Energieabhängige Ionentransporter halten Konzentrationsgradienten aufrecht. Während der Erzeugung von Aktionspotentialen bewegen sich Natriumionen durch spannungsgesteuerte Ionenkanäle über die Membran. Die ruhende Muskelfasermembran wird hauptsächlich durch die Bewegung von Chlorid durch Chloridkanäle polarisiert und durch Bewegung von Kalium repolarisiert. Natrium-, Chlorid- und Calciumkanalopathien sind als Gruppe mit Myotonie und PP assoziiert. Die funktionellen Untereinheiten von Natrium-, Calcium- und Kaliumkanälen sind homolog. Natriumkanalopathien sind besser verstanden als Calcium- oder Chloridkanalopathien. Alle Formen der familiären PP zeigen den endgültigen mechanistischen Weg, der eine aberrante Depolarisation, Inaktivierung von Natriumkanälen und Muskelfasererregbarkeit beinhaltet.
Die Diskussion in diesem Artikel befasst sich hauptsächlich mit den Natrium-, Calcium- und Kaliumkanalopathien sowie sekundären Formen von PP. Chloridkanalopathien sind nicht mit episodischer Schwäche verbunden und werden in den Artikeln über myotone Störungen ausführlicher diskutiert.
Summary of channel dysfunction in various types of PP
Bei hyperschneller Kanalinaktivierung befinden sich Mutationen üblicherweise in den inneren Teilen von Transmembransegmenten oder in den intrazellulären Schleifen, die die Andockstellen für das schnell inaktivierende Partikel beeinflussen, wodurch die schnelle Kanalinaktivierung beeinträchtigt wird, was zu einem anhaltenden Na + -Strom führt.
Bei HypoPP-hyperpolarisationsaktiviertem Kationenleck, das dem K + -gleichrichtenden Strom entgegenwirkt, verursachen Mutationen eine äußerste Arginin- oder Lysinsubstitution.
Bei NormoPP-depolarisationsaktiviertem Kationenleck liegen Mutationen an tieferen Stellen des Spannungssensors der Domäne II am Codon R675 vor.
Ionenkanalfunktionsstörungen werden normalerweise bei normaler Erregung gut kompensiert, und zusätzliche Auslöser sind häufig erforderlich, um Muskelerregbarkeit aufgrund einer anhaltenden Membrandepolarisation zu erzeugen.
Die Glukose- und Kaliumaufnahme hat bei diesen Störungen die entgegengesetzten Auswirkungen. Bei Hyperaktivität löst die Kaliumaufnahme den Angriff aus, während Glukose ihn verbessert. Im Gegensatz dazu provoziert Glukose hypokaliämische Anfälle und Kalium ist die Behandlung für den Angriff.
Beachten Sie das Bild unten.
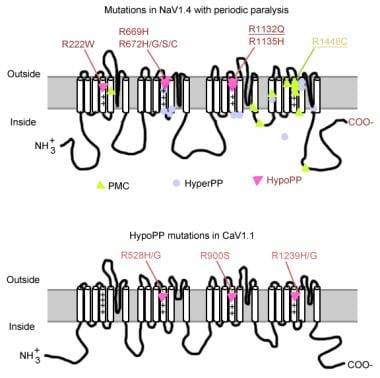 Mutationen bei periodischer Lähmung.
Mutationen bei periodischer Lähmung. Muskel-Natriumkanal-Gen
Der Natriumkanal hat eine Alpha-Untereinheit und eine Beta-Untereinheit. Die Alpha-Untereinheit des Natriumkanals ist ein 260-kd-Glykoprotein, das etwa 1800-2000 Aminosäuren umfasst. Dieser Kanal ist evolutionär von Drosophila zu Mensch hoch konserviert. Es hat 4 homologe Domänen (I-IV), die sich zu einer zentralen Pore mit jeweils 225-325 Aminosäuren falten. Jede Domäne besteht aus 6 hydrophoben Segmenten (S1-S6), die die Zellmembran durchqueren. Die Hauptfunktionen des Kanals umfassen spannungsempfindliches Gating, Inaktivierung und Ionenselektivität. Die extrazelluläre Schleife zwischen S5 und S6 taucht in die Plasmamembran ein und beteiligt sich an der Bildung der Pore. Das S4-Segment enthält an jeder dritten Position positiv geladene Aminosäuren und fungiert als Spannungssensor. Konformationsänderungen können während der Depolarisation auftreten, was zu einer Aktivierung und Inaktivierung des Kanals führt. Die Zellschleife zwischen Domäne III-S6 und Domäne IV-S1 wirkt als inaktivierendes Gate.
Der Natriumkanal hat 2 Tore (Aktivierung und Inaktivierung) und kann in 3 Zuständen existieren. Im Ruhezustand mit polarisierter Membran wird das Aktivierungstor geschlossen und das Inaktivierungstor geöffnet. Bei der Depolarisation öffnet sich das Aktivierungstor, wodurch Natriumionen durch den Ionenkanal gelangen können und auch eine Andockstelle für das Inaktivierungstor freigelegt wird. Bei fortgesetzter Depolarisation schließt sich das Inaktivierungstor, blockiert den Eintritt von Natrium in die Zelle und bewirkt, dass der Kanal in den Zustand der schnellen Inaktivierung eintritt. Durch diese Inaktivierung des Kanals kann die Membran repolarisiert werden, was zu einer Rückkehr in den Ruhezustand bei geschlossenem Aktivierungstor und geöffnetem Inaktivierungstor führt. Im Skelettmuskel von Säugetieren treten zwei Inaktivierungsprozesse auf: Bei der schnellen Inaktivierung wird das Aktionspotential beendet und es wird eine Zeitskala von Millisekunden eingehalten. Eine langsame Inaktivierung dauert Sekunden bis Minuten und kann die Population erregbarer Natriumkanäle regulieren.
Natriumkanalmutationen, die eine schnelle und langsame Inaktivierung stören, sind normalerweise mit einem Phänotyp von HyperPP und Myotonie assoziiert, wobei As-Mutationen, die eine langsame oder schnelle Inaktivierung verstärken und einen Verlust der Natriumkanalfunktion verursachen, HypoPP verursachen.
Mutationen des Natriumkanalgens (SCN4A) weisen mehrere allgemeine Merkmale auf. Die meisten Mutationen befinden sich im „inaktivierenden“ Linker zwischen den Wiederholungen III und IV, im „Voltage-Sensing“ -Segment S4 von Wiederholung IV oder an der inneren Membran, wo sie die Andockstelle für das Inaktivierungsgate beeinträchtigen könnten. Der klinische Phänotyp unterscheidet sich durch spezifische Aminosäuresubstitution und, während einige Überschneidungen zwischen hyperkalämischen PP auftreten, paramyotonia congenita (PC), und Kalium verschlimmert Myotonien (PAM), die 3 Phänotypen sind im Allgemeinen verschieden (wie unten beschrieben). Fast alle Mutantenkanäle haben eine beeinträchtigte schnelle Inaktivierung des Natriumstroms. Die meisten Patienten reagieren empfindlich auf systemisches Kalium oder kalte Temperaturen.
Es gibt zwei Populationen von Kanälen, Mutanten und Wildtypen; Die beeinträchtigte schnelle Inaktivierung führt zu einer verlängerten Depolarisation der mutierten Muskelfasermembranen und kann die 2 Hauptsymptome dieser Störungen, Myotonie und Schwäche, erklären. Bei hyperkalämischem PP tritt beim mutierten Kanal-Gating ein Funktionsgewinn auf, der zu einem erhöhten Natriumstrom führt, der den betroffenen Muskel übermäßig depolarisiert. Eine leichte Depolarisation (5-10 mV) der Myofasermembran, die durch erhöhte extrazelluläre Kaliumkonzentrationen verursacht werden kann, führt dazu, dass die Mutantenkanäle im nicht aktivierten Modus gehalten werden. Der anhaltende Natriumstrom nach innen verursacht ein wiederholtes Abfeuern der Wildtyp-Natriumkanäle, was als Steifheit (dh Myotonie) wahrgenommen wird.
Wenn eine stärkere Depolarisation (20-30 mV) vorliegt, werden sowohl normale als auch abnormale Kanäle in einem Zustand der Inaktivierung fixiert, was zu Schwäche oder Lähmung führt. Daher können subtile Unterschiede in der Schwere der Membrandepolarisation den Unterschied zwischen Myotonie und Lähmung ausmachen. Temperaturempfindlichkeit ist ein Markenzeichen von PC. Kälte verschlimmert Myotonie und induziert Schwäche. Eine Reihe von Mutationen sind mit diesem Zustand assoziiert, 3 davon an derselben Stelle (1448) im S4-Segment. Diese Mutationen ersetzen Arginin durch andere Aminosäuren und neutralisieren diese hochkonservierte positive S4-Ladung. Mutationen dieser Rückstände sind die häufigste Ursache für PC. Einige der möglichen Mechanismen, die für die Temperaturempfindlichkeit verantwortlich sind, umfassen die folgenden:
-
Die Temperatur kann die Konformationsänderung im Mutantenkanal differentiell beeinflussen.
-
Niedrigere Temperaturen können die Mutantenkanäle in einem abnormalen Zustand stabilisieren.
-
Mutationen können die Empfindlichkeit des Kanals gegenüber anderen zellulären Prozessen wie Phosphorylierung oder Second Messenger verändern.
Die meisten Fälle von hyperkalämischer PP sind auf 2 Mutationen in SCN4A, T704M und M1592V zurückzuführen. Mutationen im Natriumkanal, insbesondere an den Resten 1448 und 1313, sind für Paramyotonia congenita verantwortlich. Ein kleiner Teil der hypokaliämischen periodischen Lähmungsfälle ist mit Mutationen an den Codons 669 und 672 (HypoPP2) assoziiert. In HypoPP2 verstärken Natriumkanalmutationen die Inaktivierung, um einen Nettoverlust des Funktionsfehlers zu erzeugen.
Die normokalämische PP ähnelt sowohl der HyperPP (Kaliumsensitivität) als auch der HypoPP (Angriffsdauer) und wird durch SCN4A-Mutationen an einer tieferen Stelle des Spannungssensors DII bei Codon 675 verursacht. R675-Mutationen unterscheiden sich von HypoPP dadurch, dass diese Mutationen zu einer depolarisationsaktivierten Gating-Pore führen, die ω-Strom mit umgekehrter Spannungsabhängigkeit erzeugt, da diese Stelle bei stärkerer Depolarisation extrazellulären Stellen ausgesetzt ist.
Kalziumkanalgen
Das Kalziumkanalgen (CACNL1A3) ist ein Komplex aus 5 Untereinheiten (Alpha-1, Alpha-2, Beta, Gamma und Delta). Der Dihydropyridin (DHP) -Rezeptor des Skelettmuskels befindet sich hauptsächlich in der transversalen tubulären Membran. Die Alpha-1-Untereinheit hat Bindungsstellen für DHP-Medikamente und leitet den langsamen L-Typ-Calciumstrom. Es beteiligt sich auch an der Anregungs-Kontraktions-Kopplung (EC) und wirkt als Spannungssensor durch seine Verknüpfung mit dem Ryanodinrezeptor des sarkoplasmatischen Retikulums (dh dem Calciumfreisetzungskanal). Jede Änderung des Membranpotentials ist mit der intrazellulären Calciumfreisetzung verbunden, was eine EC-Kopplung ermöglicht. Punktmutationen in der DHP-Rezeptor / Calciumkanal-Alpha-1-Untereinheit verursachen hypokaliämische PP (HypoPP1). Zwei Mutationen des CACNA1S-Gens, R528H und R1239H, sind für die meisten Fälle von hypokaliämischer PP verantwortlich.
Die physiologischen Grundlagen der Krankheit sind noch nicht verstanden, sind aber eher auf ein Versagen der Erregung als auf ein Versagen der EC-Kopplung zurückzuführen. Eine Hypokaliämie-induzierte Depolarisation kann jedoch die Calciumfreisetzung verringern und die Spannungssteuerung des Kanals direkt oder indirekt durch Inaktivierung des Natriumkanals beeinflussen. Insulin und Adrenalin können in ähnlicher Weise wirken. Mutationen des Calciumkanalgens haben einige Ähnlichkeiten mit SCN4A-Mutationen. Mutationen modifizieren die Kanalinaktivierung, aber nicht die spannungsabhängige Aktivierung. Aufnahmen von Myotube-Kulturen von betroffenen Patienten zeigten eine 30% ige Reduktion des DHP-sensitiven L-Typ-Calciumstroms. Kanäle werden bei niedrigen Membranpotentialen inaktiviert.
Calciumkanalmutationen verursachen einen Funktionsverlust, der sich in einer verringerten Stromdichte und einer langsameren Inaktivierung äußert. Wie diese Inaktivierung mit Hypokaliämie-induzierten Anfällen zusammenhängt, ist nicht verstanden. Zumindest bei der R528H-Mutation tritt eine mögliche sekundäre Kanalopathie auf, die mit einer Verringerung des ATP-sensitiven Kaliumstroms aufgrund einer veränderten Calciumhomöostase verbunden ist. Die niedrigeren Ströme, die mit CACNL1A3-Mutationen assoziiert sind, könnten die intrazelluläre Calciumhomöostase geringfügig verändern, was die Eigenschaften und Expression von K + -Kanälen beeinflussen könnte, insbesondere KATP (ATP-sensitiver Kaliumkanal), der zur gleichen Kanalklasse gehört. Insulin wirkt auch bei Unterzuckerung, indem es diesen nach innen gerichteten Gleichrichter-K + -Strom reduziert.
Der Ladungsverlust des Spannungssensors macht die meisten Fälle von HypoPP aus. Natrium- und Calciumkanäle weisen homologe porenbildende Alfa-Untereinheiten auf. Punktmutationen in CACNL1A3 und SCN4A beeinflussen die Reste in den S4-Spannungssensoren dieser Kanäle. Argininmutationen in S4-Segmenten sind für 90% der HypoPP-Fälle verantwortlich.
Der Ladungsverlust des Spannungssensors macht die meisten Fälle von HypoPP aus. Natrium- und Calciumkanäle haben homologe porenbildende α-Untereinheiten. Fast alle Mutationen in Cav1.1 (HypoPP-1) und Nav1.4 (HypoPP-2) neutralisieren eine positiv geladene Aminosäure in einem der äußersten Arginine oder Lysine von Spannungssensoren. Das Nav1.4 Mutationen befinden sich am häufigsten in den Spannungssensoren der Wiederholungen I, II und III und verursachen ein Kationenleck.
Die Substitution von äußerstem Arginin durch eine kleinere Aminosäure wie Glycin öffnet einen leitenden Weg bei hyperpolarisiertem Potential, was zu einem nach innen gerichteten Kationenstrom (Kationenstrom oder ω-Strom zur Unterscheidung von (ω–) durch ionenleitende Pore, ist ein hyperpolarisationsaktivierter Strom von einwertigen Kationen durch S4-Gating-Poren, die gleichrichtenden K + -Strömen entgegenwirken), der das Ruhepotential depolarisiert oder destabilisiert.
Das S4-Segment bewegt sich während der Depolarisation nach außen und schließt den leitenden Pfad. Muskelfasern mit schweren Spannungssensormutationen werden nicht nur während der Hypokaliämie depolarisiert, sondern auch bei Kaliumspiegeln im normalen Bereich, was interiktale und permanente Schwäche erklärt. Schwere Myopathie mit fettem Ersatz von Muskelgewebe wird häufig bei Patienten mit Cav1.1 R1239H (DIV-Mutationen) gefunden.
Glukokortikosteroide verursachen HypoPP, indem sie die durch Insulin und Amylin vermittelte Na + K + ATPase stimulieren.
Kaliumkanalgen
Die nach innen gerichtete Rektifikation ist eine wichtige Eigenschaft von Kir-Kanälen. Die Rektifikation beinhaltet eine spannungsabhängige Leitungsporenblockade der Pore mit Polyaminen und Mg ++ während der Depolarisation, und diese Blockierung wird während des Potentialgradienten während der Hyperpolarisation entfernt. Kaliumkanalmutationen treten beim Andersen-Tawil-Syndrom und bei thyreotoxischen PP auf.
Der Dreiklang aus dysmorphen Merkmalen, periodischer Lähmung und Herzrhythmusstörungen charakterisiert das Andersen-Tawil-Syndrom. Dieses Syndrom ist mit Mutationen im KCNJ2-Gen assoziiert. Das KCNJ2-Gen kodiert für den nach innen korrigierenden Kaliumkanal Kir2.1. Es wird berichtet, dass Kaliumkanalmutationen in KCNE3 hypokaliämische PP verursachen, dies wurde jedoch nicht belegt.
Mutationen in Kir2.6 verursachen eine Anfälligkeit für thyreotoxische PP. Episodische Schwäche in thyreotoxischen PP gesehen ist ähnlich wie bei HypoPP und Andersen-Tawil-Syndrom gesehen. Diese Störung ist am häufigsten bei Asiaten und lateinamerikanischen Männern. Thyreotoxikose ist eine genetische Störung, die durch Thyreotoxikose entlarvt wird. Kir2.6 wird hauptsächlich im Skelettmuskel exprimiert. Trijodthyronin verstärkt die KCNJ18-Transkription, was zu einer verstärkten Expression von Kir2.6 führen kann. PKC wird während der Thyreotoxikose aufgrund des erhöhten PIP2-Umsatzes aktiviert und Kir-Kanäle interagieren während des normalen Gating direkt mit PIP2. Beim Andersen-Tawil-Syndrom ist die PIP2-Affinität vermindert. Bei thyreotoxischer PP verändert keine der Mutationen die Kir2.6-Rektifikation.