Atrofia Muscular espinal
Causas / Herencia
¿Qué causa la atrofia muscular espinal (AME)?
La AME se caracteriza por la pérdida de neuronas motoras, células nerviosas en la médula espinal. Se clasifica como una enfermedad de la neurona motora.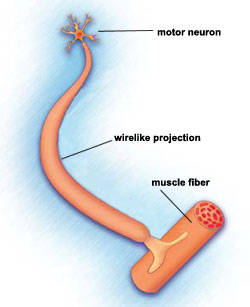
¿Cuáles son las causas genéticas de la AME?
La forma más común de AME (tipos 1-4) es causada por un defecto (mutación) en el gen SMN1 en el cromosoma 5. (Las personas tienen dos genes SMN1, uno en cada cromosoma 5). En el 94% de todos los casos de AME, esta mutación implica una deleción en un segmento conocido como exón 7. Esta área se encuentra en el brazo largo del cromosoma 5, en la región 5q13.2 (los cromosomas tienen dos «brazos»: uno corto, identificado por la letra «p», y uno largo, identificado por la letra «q»).
Una mutación en el gen SMN1 conduce a una deficiencia de una proteína de la neurona motora llamada SMN, que significa «supervivencia de la neurona motora».»Como su nombre lo indica, esta proteína es responsable de la expresión génica necesaria para la función normal de la neurona motora.
Más raramente, una mutación en un gen del cromosoma X llamado UBE1 causa AME ligada al cromosoma X. El gen UBE1 lleva instrucciones para la enzima 1 activadora de ubiquitina, que normalmente ayuda a unir una etiqueta molecular a las proteínas para marcarlas para su destrucción.
Se ha encontrado que los defectos en el gen citoplasmático de la cadena pesada 1 de la dineína 1 (DYNC1H1) en el cromosoma 14 conducen a otra forma rara de AME llamada AME-LED.
¿Qué causa la amplia variación en la gravedad de los síntomas observada en la AME?
Normalmente, los genes SMN1 producen proteína SMN de longitud completa y completamente funcional. Pero cuando el gen SMN1 tiene mutaciones, como en la forma de SMA relacionada con el cromosoma 5, se producen niveles insuficientes de proteína SMN.
Un gen vecino en el cromosoma 5, llamado SMN2, también produce proteína SMN. La mayor parte de la proteína elaborada a partir de instrucciones transportadas por los genes SMN2 no es funcional, pero un pequeño porcentaje, alrededor del 10 al 15%, es funcional.
Las personas pueden tener múltiples copias del gen SMN2. Normalmente, el número varía entre cero y ocho copias. En la forma de AME relacionada con el cromosoma 5, cuantas más copias del gen SMN2 tenga una persona, más funcional estará disponible la proteína SMN. Como resultado, es probable que el curso de la enfermedad sea más leve. Tener tres o más copias del gen SMN2 se asocia con una manifestación de la enfermedad menos grave.
Las pruebas genéticas pueden indicar cuántos genes SMN2 tiene una persona y predecir aproximadamente el curso de la AME que es probable que se produzca.
La gravedad de la AME también puede depender de los modificadores de la enfermedad, que no causan la enfermedad, pero pueden afectar (modificar) el inicio y la gravedad al influir en varias vías biológicas. Los niveles de proteína plastina 3 y de proteína ZPR1 se han identificado como modificadores de la AME relacionada con la NSM y podrían convertirse en dianas terapéuticas. Además, las pruebas para estos niveles de proteínas podrían ayudar a predecir la gravedad de la enfermedad, y la comprensión de las actividades de estas proteínas podría arrojar nueva luz sobre los procesos de la enfermedad.

La información genética pasa de su forma de almacenamiento como ADN a un conjunto de instrucciones conocidas como ARN, a partir de las cuales se fabrican moléculas de proteínas. La mayoría de las instrucciones de ARN del gen SMN1 indican a la célula que produzca proteína SMN de longitud completa. La mayoría de las instrucciones del gen SMN2 indican a la célula que produzca proteína SMN corta.
¿Cuál es el patrón de herencia de SMA?
La AME relacionada con el cromosoma 5 (tipos 1 a 4) sigue un patrón hereditario conocido como autosómico recesivo. (Los autosomas son los cromosomas numerados, es decir, todos los cromosomas excepto el X y el Y, que determinan el género.)
Las enfermedades recesivas requieren dos defectos genéticos, generalmente uno de cada padre, pero ocasionalmente uno de uno de los padres y uno que ocurre cuando se forma un feto. Se dice que las personas que tienen un solo defecto genético para una enfermedad recesiva son portadoras y, por lo general, no muestran síntomas. A menudo, una familia no tiene idea de que algunos miembros son portadores hasta que un niño nace con un trastorno recesivo.
Si ambos padres son portadores del defecto del gen cromosoma 5, el riesgo de que cada embarazo produzca un hijo con la enfermedad es del 25%. Este riesgo no cambia sin importar cuántos hijos tenga una pareja.
Las pruebas genéticas para la AME relacionada con el cromosoma 5 están disponibles para las personas sospechosas de tener la enfermedad, incluidos los bebés no nacidos, y para los portadores de la enfermedad. Las pruebas genéticas se están expandiendo y cambiando rápidamente, pero sus implicaciones pueden ser complejas. Lo mejor es hablar con un asesor genético antes de emprender la prueba. (Se puede obtener una referencia de asesoramiento genético a través de su Centro de Atención de MDA o de su médico de atención primaria).
Es imperativo diagnosticar la AME lo antes posible, idealmente antes de la aparición de los síntomas, porque el retraso en el tratamiento puede empeorar el curso de la enfermedad y crear daño permanente a las neuronas motoras. La mejor manera de identificar la AME antes de que aparezcan los síntomas es examinar a todos los recién nacidos para detectar la eliminación del exón 7 de SMN1. Dado que Spinraza (nusinersen), una terapia modificadora de la enfermedad, fue aprobada en 2016 por la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) y el tratamiento temprano mostró resultados positivos, el secretario del Departamento de Salud de los Estados Unidos aprobó la adición de SMA a los paneles de detección de recién nacidos.1 visit para obtener más información, visite el HIPERVÍNCULO «https://strongly.mda.org/sma-added-national-list-disorders-to-screen-for…»SMA Agregado a la Lista Nacional de Trastornos para Detectar al Nacer.
La AME ligada al cromosoma X se hereda a través del cromosoma X. Las mujeres tienen dos cromosomas X, y las que tienen un defecto genético en un cromosoma X generalmente se consideran portadoras de una enfermedad ligada al cromosoma X. Los hombres, sin embargo, no tienen un segundo X para protegerlos de los efectos completos de un defecto genético en el cromosoma X y mostrar los efectos completos de dicho defecto.
Además, la AME puede ser causada por mutaciones en el gen DYNC1H1 en el cromosoma 14. Esta forma es predominantemente hereditaria, lo que significa que solo una mutación del gen DYNC1H1, heredada de uno de los padres, es suficiente para causar la enfermedad.
Para obtener más información sobre la genética de la AME y las pruebas genéticas para esta enfermedad, consulte también:
- Seminario Web de Asesoramiento Genético de MDA, Respuestas a Preguntas Clave, Quest News En Línea, Febrero. 28, 2012
- Facts About Genetics and Neuromuscular Diseases, MDA, Diciembre de 2009
- The Genie’s Out of the Bottle: Genetic testing in the 21st century, Quest Magazine (MDA), noviembre de 2008
- El dolor y la Promesa del Diagnóstico Genético Prenatal y Neonatal, Quest Magazine (MDA), julio de 2007