Parálisis Periódica
Una clasificación clínicamente útil de las parálisis periódicas primarias, que se muestra en la Tabla 1, incluye las formas hipocalémica, hipercalémica y paramiotónica.
Cuadro 1. Primaria Parálisis Periódica (modificado de Jurkat-Rott y Lehmann-Horn ) (Tabla Abierta en una ventana nueva)
|
la Enfermedad |
Gen |
la Proteína |
la Herencia |
la Mutación |
|
HyperPP |
SCN4A |
Nav1.4 |
Dominante |
Fina |
|
NormoPP |
Fina (ω-poros) |
|||
|
Paramyotoniacongenita |
Fina |
|||
|
HypoPP Tipo II |
Fina (ω-poros) |
|||
|
HypoPP el Fin DE |
CACNA1S |
Cav1.1 |
Dominante |
Ganancia (ω-poros) |
|
ThyrotoxicPP |
KCNJ18 |
Kir2.18 |
Dominante |
la Pérdida de |
|
síndrome de Andersen-Tawil |
KCNJ2 |
Kir2.1 |
Dominante |
la Pérdida de |
La base fisiológica de la debilidad flácida es inexcitabilidad de la membrana muscular (es decir, el sarcolema). La alteración de los niveles séricos de potasio no es el defecto principal en el PP primario; el metabolismo alterado del potasio es el resultado del PP. En la PP primaria y tirotóxica, la parálisis flácida ocurre con cambios relativamente pequeños en el nivel sérico de potasio, mientras que en la PP secundaria, los niveles séricos de potasio son marcadamente anormales.
Ningún mecanismo es responsable de este grupo de trastornos. Por lo tanto, son heterogéneos, pero comparten algunos rasgos comunes. La debilidad generalmente es generalizada, pero puede estar localizada. La musculatura craneal y los músculos respiratorios generalmente se salvan. Los reflejos de estiramiento están ausentes o disminuidos durante los ataques. Las fibras musculares son eléctricamente inexcitables durante los ataques. La fuerza muscular es normal entre ataques, pero, después de unos años, se desarrolla cierto grado de debilidad fija en ciertos tipos de PP (especialmente PP primario). Todas las formas de PP primaria (excepto la miotonía congénita de Becker ) son hereditarias autosómicas dominantes o esporádicas (lo más probable es que surjan de mutaciones puntuales).
Los canales iónicos sensibles al voltaje regulan estrechamente la generación de potenciales de acción (alteraciones breves y reversibles de la tensión de las membranas celulares). Estos son canales iónicos permeables de forma selectiva y variable. Los transportadores de iones dependientes de energía mantienen gradientes de concentración. Durante la generación de potenciales de acción, los iones de sodio se mueven a través de la membrana a través de canales iónicos dependientes de voltaje. La membrana de fibra muscular en reposo se polariza principalmente por el movimiento del cloruro a través de los canales de cloruro y se repolariza por el movimiento del potasio. Las canalopatías de sodio, cloruro y calcio, como grupo, se asocian con miotonía y PP. Las subunidades funcionales de los canales de sodio, calcio y potasio son homólogas. Las canalopatías de sodio se entienden mejor que las canalopatías de calcio o cloruro. Todas las formas de PP familiar muestran la vía mecánica final que implica despolarización aberrante, inactivación de los canales de sodio e inexcitabilidad de la fibra muscular.
La discusión en este artículo aborda principalmente las canalopatías de sodio, calcio y potasio, así como las formas secundarias de PP. Las canalopatías de cloruro no están asociadas con debilidad episódica y se discuten con más detalle en los artículos sobre trastornos miotónicos.
Resumen de la disfunción del canal en varios tipos de PP
Con la inactivación rápida del canal de HyperPP, las mutaciones generalmente se sitúan en las partes internas de los segmentos transmembranarios o en los bucles intracelulares que afectan a los sitios de acoplamiento de la partícula de inactivación rápida, lo que perjudica la inactivación rápida del canal que conduce a una corriente persistente de Na+.
Con HipoPP, corriente rectificadora K+ contrarrestadora de fugas de cationes activadas por hiperpolarización, las mutaciones causan sustitución externa de arginina o lisina.
Con fuga de cationes activada por despolarización de NormoPP, las mutaciones se encuentran en ubicaciones más profundas del sensor de voltaje del dominio II en el codón R675.
La disfunción del canal iónico generalmente se compensa bien con la excitación normal, y a menudo se necesitan desencadenantes adicionales para producir inexcitabilidad muscular debido a la despolarización sostenida de la membrana.
La ingesta de glucosa y potasio tiene los efectos opuestos en estos trastornos. En HyperPP, la ingesta de potasio desencadena el ataque, mientras que la glucosa lo mejora. En contraste, la glucosa provoca hipocaliémica ataques y potasio es el tratamiento para el ataque.
Observe la imagen de abajo.
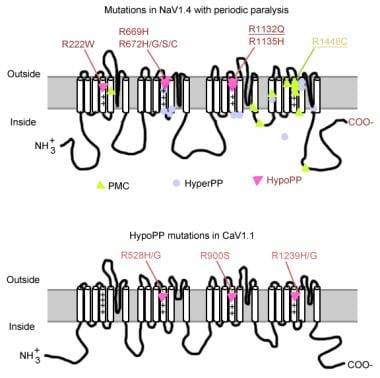 Mutaciones en parálisis periódica.
Mutaciones en parálisis periódica. Gen del canal de sodio muscular
El canal de sodio tiene una subunidad alfa y una subunidad beta. La subunidad alfa del canal de sodio es una glicoproteína de 260 kd que comprende alrededor de 1800-2000 aminoácidos. Este canal está altamente conservado evolutivamente de Drosophila a humano. Tiene 4 dominios homólogos (I-IV) que se pliegan para formar un poro central, cada uno con 225-325 aminoácidos. Cada dominio consta de 6 segmentos hidrofóbicos (S1-S6) que atraviesan la membrana celular. Las funciones principales del canal incluyen la activación sensible a la tensión, la inactivación y la selectividad iónica. El bucle extracelular entre S5 y S6 se sumerge en la membrana plasmática y participa en la formación del poro. El segmento S4 contiene aminoácidos cargados positivamente en cada tercera posición y funciona como un sensor de voltaje. Los cambios de conformación pueden ocurrir durante la despolarización, lo que resulta en la activación e inactivación del canal. El bucle celular entre el dominio III-S6 y el dominio IV-S1 actúa como puerta inactivadora.
El canal de sodio tiene 2 puertas (activación e inactivación) y puede existir en 3 estados. En reposo con la membrana polarizada, la puerta de activación se cierra y la puerta de inactivación se abre. Con la despolarización, la puerta de activación se abre, permitiendo que los iones de sodio pasen a través del canal iónico y también exponiendo un sitio de acoplamiento para la puerta de inactivación. Con la despolarización continua, la puerta de inactivación se cierra, bloqueando la entrada de sodio en la célula y haciendo que el canal entre en el estado de inactivación rápida. Esta inactivación del canal permite que la membrana se repolarice, lo que resulta en un retorno al estado de reposo con la puerta de activación cerrada y la puerta de inactivación abierta. Dos procesos de inactivación ocurren en el músculo esquelético de mamíferos: La inactivación rápida implica terminar el potencial de acción y actúa en una escala de tiempo de milisegundos. La inactivación lenta tarda de segundos a minutos y puede regular la población de canales de sodio excitables.
Las mutaciones en los canales de sodio que interrumpen la inactivación rápida y lenta generalmente se asocian con un fenotipo de HiperPP y miotonía, donde las mutaciones que mejoran la inactivación lenta o rápida que producen pérdida de la función de los canales de sodio causan HipoPP.
Las mutaciones del gen del canal de sodio (SCN4A) tienen varias características generales. La mayoría de las mutaciones se encuentran en el enlazador» inactivador «entre las repeticiones III y IV, en el segmento» sensor de voltaje » S4 de la repetición IV o en la membrana interna donde podrían dañar el sitio de acoplamiento para la puerta de inactivación. El fenotipo clínico difiere en función de la sustitución de aminoácidos específicos y, aunque se puede producir cierta superposición entre PP hipercalémico, paramiotonía congénita (PC) y miotonias agravadas por potasio (PAM), los 3 fenotipos son generalmente distintos (como se describe a continuación). Casi todos los canales mutantes han alterado la inactivación rápida de la corriente de sodio. La mayoría de los pacientes son sensibles al potasio sistémico o a la temperatura fría.
Existen dos poblaciones de canales, mutantes y de tipo salvaje; el deterioro de la inactivación rápida resulta en una despolarización prolongada de las membranas de fibra muscular mutantes y puede explicar los 2 síntomas cardinales de estos trastornos, miotonía y debilidad. En la PP hipercalémica, se produce una ganancia de función en la apertura de canales mutantes, lo que resulta en un aumento de la corriente de sodio que despolariza excesivamente el músculo afectado. La despolarización leve (5-10 mV) de la membrana de miofibras, que puede ser causada por el aumento de las concentraciones extracelulares de potasio, da lugar a que los canales mutantes se mantengan en el modo no inactivado. La corriente persistente de sodio hacia adentro causa el disparo repetitivo de los canales de sodio de tipo salvaje, que se percibe como rigidez (es decir, miotonía).
Si se presenta una despolarización más severa (20-30 mV), los canales normales y anormales se fijan en un estado de inactivación, causando debilidad o parálisis. Por lo tanto, las diferencias sutiles en la gravedad de la despolarización de la membrana pueden marcar la diferencia entre la miotonía y la parálisis. La sensibilidad a la temperatura es un sello distintivo de la PC. El frío exacerba la miotonía e induce debilidad. Hay varias mutaciones asociadas con esta afección, 3 de ellas en el mismo sitio (1448) en el segmento S4. Estas mutaciones reemplazan la arginina con otros aminoácidos y neutralizan esta carga positiva S4 altamente conservada. Las mutaciones de estos residuos son la causa más común de PC. Algunos de los posibles mecanismos responsables de la sensibilidad a la temperatura incluyen los siguientes:
-
la Temperatura puede afectar diferencialmente el cambio conformacional en el canal mutante.
-
Temperaturas más bajas pueden estabilizar los canales mutantes en un estado anormal.
-
Las mutaciones pueden alterar la sensibilidad del canal a otros procesos celulares, como la fosforilación o segundos mensajeros.
La mayoría de los casos de PP hipercalémica se deben a 2 mutaciones en SCN4A, T704M y M1592V. Las mutaciones en el canal de sodio, especialmente en los residuos 1448 y 1313, son responsables de la paramiotonía congénita. Una pequeña proporción de casos de parálisis periódica hipocaliémica están asociados con mutaciones en los codones 669 y 672 (HipoPP2). En HypoPP2, las mutaciones del canal de sodio aumentan la inactivación para producir una pérdida neta de defecto funcional.
La PP normocalémica se asemeja tanto a la HiperPP (sensibilidad al potasio) como a la HipoPP (duración de los ataques) y es causada por mutaciones en SCN4A en una ubicación más profunda del sensor de voltaje DII en el codón 675. Las mutaciones R675 difieren de HypoPP en que estas mutaciones resultan en la generación de poros de compuerta activados por despolarización que generan corriente ω con dependencia de voltaje invertida, ya que este sitio está expuesto a sitios extracelulares en una despolarización más fuerte.
Gen del canal de calcio
El gen del canal de calcio (CACNL1A3) es un complejo de 5 subunidades (alfa-1, alfa-2, beta, gamma y delta). El receptor de dihidropiridina (DHP) del músculo esquelético se encuentra principalmente en la membrana tubular transversal. La subunidad alfa-1 tiene sitios de unión para los fármacos DHP y conduce la corriente lenta de calcio de tipo L. También participa en el acoplamiento de excitación-contracción (CE) y actúa como un sensor de voltaje a través de su enlace con el receptor de rianodina del retículo sarcoplásmico (es decir, canal de liberación de calcio). Cualquier cambio en el potencial de membrana está relacionado con la liberación de calcio intracelular, lo que permite el acoplamiento de la CE. Las mutaciones puntuales en el receptor DHP/subunidad alfa-1 del canal de calcio causan PP hipocalémico (HipoPP1). Dos mutaciones del gen CACNA1S, R528H y R1239H, son responsables de la mayoría de los casos de PP hipocalémico.
La base fisiológica de la enfermedad todavía no se comprende, pero es más probable que se deba a un fallo de la excitación que a un fallo del acoplamiento de la CE. Sin embargo, la despolarización inducida por hipopotasemia puede reducir la liberación de calcio, afectando el control de voltaje del canal directa o indirectamente a través de la inactivación del canal de sodio. La insulina y la adrenalina pueden actuar de una manera similar. Las mutaciones del gen del canal de calcio tienen algunas similitudes con las mutaciones de SCN4A. Las mutaciones modifican la inactivación del canal, pero no la activación dependiente del voltaje. Las grabaciones de cultivos de miotubos de pacientes afectados revelaron una reducción del 30% en la corriente de calcio de tipo L sensible al DHP. Los canales se inactivan a bajos potenciales de membrana.
Las mutaciones en los canales de calcio causan una pérdida de función que se manifiesta como una densidad de corriente reducida y una inactivación más lenta. No se entiende cómo se relaciona esta inactivación con los ataques inducidos por hipopotasemia. Al menos en la mutación R528H, se produce una posible canalopatía secundaria, vinculada a una reducción en la corriente de potasio sensible al ATP de la homeostasis de calcio alterada. Las corrientes inferiores asociadas con mutaciones en CACNL1A3 podrían alterar ligeramente la homeostasis intracelular del calcio, lo que podría afectar las propiedades y la expresión de los canales K+, en particular el KATP (canal de potasio sensible a ATP) perteneciente a la clase de canales rectificadores internos. La insulina también actúa en la HipoPP reduciendo esta corriente K + del rectificador interno.
Cuentas de pérdida de carga del sensor de voltaje para la mayoría de los casos de HipoPP. Los canales de sodio y calcio tienen subunidades alfa homólogas formadoras de poros. Las mutaciones puntuales en CACNL1A3 y SCN4A afectan a los residuos argentinos en los sensores de tensión S4 de estos canales. Las mutaciones de arginina en segmentos S4 son responsables del 90% de los casos de HipoPP.
Cuentas de pérdida de carga del sensor de voltaje para la mayoría de los casos de HipoPP. Los canales de sodio y calcio tienen subunidades α homólogas formadoras de poros. Casi todas las mutaciones en Cav1.1 (HipoPP-1) y Nav1.4 (HipoPP-2) neutralizan un aminoácido cargado positivamente en una de las argininas o lisinas más externas de los sensores de voltaje. El Nav1.4 las mutaciones se localizan más comúnmente en los sensores de voltaje de las repeticiones I, II y III, causando una fuga de cationes.
La sustitución de la arginina más externa por un aminoácido más pequeño, como la glicina, abre una vía conductora en potencial hiperpolarizado, lo que resulta en una corriente catiónica interna (fuga de cationes o corriente ω para distinguirla de (ω-) a través de poro conductor de iones, es una corriente activada por hiperpolarización de cationes monovalentes a través de corrientes K+ rectificadoras contrarrestantes de poros S4) despolarizando o desestabilizando el potencial de reposo.
El segmento S4 se mueve hacia afuera durante la despolarización cerrando la vía conductora. Las fibras musculares con mutaciones graves del sensor de voltaje se despolarizan no solo durante la hipopotasemia, sino también a niveles de potasio en el rango normal, lo que explica la debilidad interictal y permanente. La miopatía grave con reemplazo graso de tejido muscular se encuentra comúnmente en pacientes con Cav1.1 R1239H (mutaciones DIV).
Los glucocorticosteroides causan HipoPP al estimular la Na + K + ATPasa mediada por insulina y amilina.
Gen del canal de potasio
La rectificación interna es una propiedad importante de los canales Kir. La rectificación implica el bloqueo de poros de conducción dependiente del voltaje con poliaminas y Mg++ durante la despolarización, y este bloqueo se elimina durante el gradiente potencial durante la hiperpolarización. Se observan mutaciones en los canales de potasio en el síndrome de Andersen-Tawil y en la PP tirotóxica.
La tríada de características dismórficas, parálisis periódica y arritmias cardíacas caracteriza al síndrome de Andersen-Tawil. Este síndrome se asocia con mutaciones en el gen KCNJ2. El gen KCNJ2 codifica el canal de potasio rectificador hacia adentro Kir2.1. Se ha informado que las mutaciones de los canales de potasio en KCNE3 causan PP hipocalémico, pero esto no se ha demostrado.
Las mutaciones en Kir2. 6 causan susceptibilidad al PP tirotóxico. La debilidad episódica observada en la PP tirotóxica es similar a la observada en la HipoPP y el síndrome de Andersen-Tawil. Este trastorno es más prevalente en los hombres asiáticos y latinoamericanos. La PP tirotóxica es un trastorno genético desenmascarado por la tirotoxicosis. Kir2. 6 se expresa principalmente en el músculo esquelético. La triyodotironina mejora la transcripción de KCNJ18, lo que puede impulsar la expresión mejorada de Kir2.6. La PKC se activa durante la tirotoxicosis debido al aumento de la rotación de PIP2 y los canales Kir interactúan directamente con PIP2 durante la apertura normal. En el síndrome de Andersen-Tawil, hay una disminución de la afinidad por el PIP2. En PP tirotóxicos, ninguna de las mutaciones altera la rectificación de Kir2. 6.