Reporte de caso de Ginecología y Obstetricia
Palabras Clave
Tetra-amelia; Malformación; Genotipo; Fenotipo.
Introducción
Las anomalías de las extremidades constituyen un grupo importante de patologías congénitas caracterizadas por hipoplasia o aplasia completa de uno o más huesos de las extremidades. Las anomalías de las extremidades de todo tipo ocurren en aproximadamente 1 de cada 1.300 a 2.000 nacimientos. Estas anomalías de las extremidades pueden aislarse o asociarse con otras anomalías . El síndrome de tetraamelia es poco frecuente y persisten áreas grises.
Reportamos dos casos de tetra-amelia en una maternidad de nivel II en Dakar (Senegal) con ser similar a tetraamelia-1 (cromosoma 17q21), tetraamelia-2 (cromosoma 8q23) y síndrome de Robert (cromosoma 8p21). Esto ilustra la dificultad de correlacionar fenotipo y genes involucrados.
Reportes de casos
Caso 1
Ms AD fue una madre de 44 años de edad remitida a nuestro departamento a las 36 semanas de gestación con preeclampsia severa y anomalías fetales. Tenía cinco para sin antecedentes de anomalías fetales. Ahora no fumaba y nunca había fumado ni bebido alcohol. No había estado expuesta al tabaquismo pasivo. Estaba en un matrimonio consanguíneo de tercer grado para todos sus hijos. La Sra. AD había dado negativo para hepatitis B, VIH y sífilis. Estaba protegida del virus de la rubéola y no había estado expuesta previamente al Toxoplasma gondii. La monitorización ecográfica realizada a las 33 y 35 semanas de gestación recuperó oligoamniosis e hidrocefalia, así como agenesia de las extremidades. Las recetas durante el embarazo incluyeron la administración de hierro y ácido fólico, así como la administración de sulfadoxina pirimetamina. Este último se prescribió a las 18 semanas y luego a las 26 semanas como parte de la política de profilaxis antipalúdica para mujeres embarazadas. La altura sinfiseal-fundal medía 28 cm. Debido a las características severas de preeclampsia, fue hospitalizada de inmediato y observada en una unidad de trabajo de parto y parto. Luego, inicialmente, recibió sulfato de magnesio intravenoso para prevenir la eclampsia y medicamentos antihipertensivos para mantener la presión arterial sistólica por debajo de 160 mmHg y la presión arterial diastólica por debajo de 105 mmHg.
Se tomó la decisión de realizar una cesárea inmediata. Se extrajeron 2.150 gramos nacidos vivos que posteriormente murieron en 10 minutos. Antes de la muerte, el cuerpo presentaba movimientos rastreros. Se identificaron varias anomalías externas (Figura 1), incluida la agenesia completa de las cuatro extremidades, hidrocefalia con un perímetro cefálico de 39 cm. En la cara, había hipertelorismo con un coloboma de los párpados, exoftalmos leves y aniridia. La boca era similar a una V invertida sin una clara delimitación de los labios y la nariz era rudimentaria. El recién nacido carecía de epidermis (pelo y cejas). Las orejas se redujeron a bocetos que parecían hendiduras. El cuello era corto. El tronco se redujo a una estructura cónica de 26 cm de largo con un cordón umbilical en el extremo inferior. El pecho era estrecho. Justo debajo del ombligo, el tronco ensanchado hacia atrás estaba presente en el suelo del polo caudal, una línea media con recesiones y un brote que puede corresponder a un falo de tipo indeterminado. Se observó agenesia de pelvis, genitales e imperforación anal. No se ha realizado patología fetal. Sin embargo, la muerte dentro de los 10 minutos posteriores al parto y la apariencia cónica del tórax pueden sugerir anomalías pulmonares.
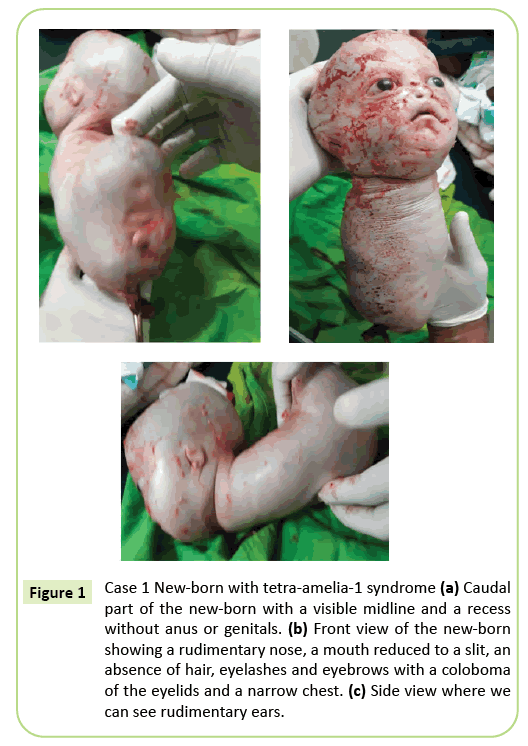
Figura 1: Caso 1 Recién nacido con síndrome tetra-amelia-1 (a) Parte caudal del recién nacido con una línea media visible y un hueco sin ano ni genitales. b) Vista frontal del recién nacido que muestra una nariz rudimentaria, una boca reducida a una hendidura, ausencia de cabello, pestañas y cejas con un coloboma de los párpados y un pecho estrecho. (c) Vista lateral donde podemos ver orejas rudimentarias.
Caso 2
El segundo caso fue un primigravida de 22 años remitido a nuestro centro para ecografía a las 37 semanas de gestación. No estaba en un matrimonio consanguíneo. Había dado negativo para hepatitis B, VIH y sífilis. No le hicieron pruebas de toxoplasmosis y rubéola. No se realizó un monitoreo por ultrasonido durante su embarazo. El examen clínico fue consistente con retraso en el crecimiento fetal (altura del fondo uterino: 26 cm). Los hallazgos ecográficos mostraron que el húmero estaba distorsionado midiendo 23,9 mm, lo que corresponde a 17 semanas de gestación. Había agenesia del fémur. Las alas ilíacas eran visibles en el ultrasonido. No se identificaron anomalías pulmonares o cardíacas. Se inició la entrega. El recién nacido tenía un fenotipo femenino con una puntuación de Apgar de 9 a los 5 minutos. La morfología de la cabeza y el tronco no tenía particularidad. Las extremidades superiores se redujeron a dos muñones de 3 cm de largo. Se observó agenesia completa de los 2 miembros inferiores. Se trataba de una anomalía simétrica (Figura 2).
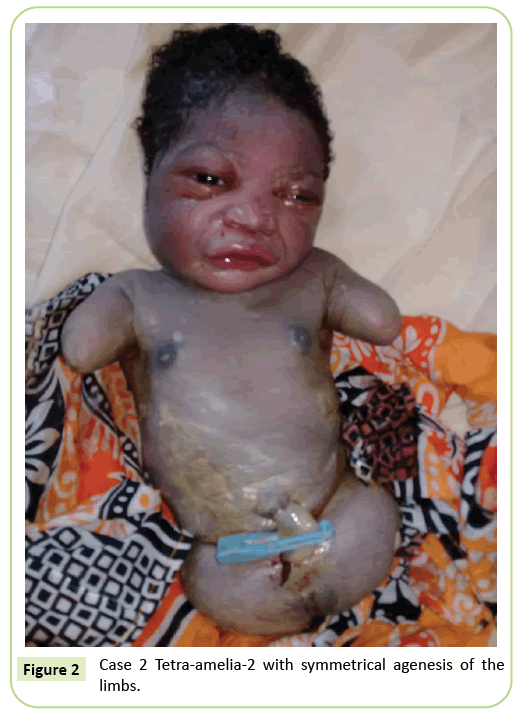
Figura 2: Caso 2 Tetra-amelia-2 simétrica con agenesia de las extremidades.
Discusión
Bermejo-Sánchez et al. en 2011 se describió la epidemiología de la amelia congénita utilizando datos recopilados de 20 programas de vigilancia de anomalías congénitas, de todos los continentes excepto África, entre 1968 y 2006. En total, se identificaron 326 casos de amelia entre 23.110.591 nacidos vivos, mortinatos y abortos. La prevalencia fue de 1.41/100,000 .
Tetra-amelia se refiere a la ausencia completa de las extremidades y ocurre más raramente. Hasta donde sabemos, tetra-amelia-1 se describe en 7 familias. Parece seguir una herencia autosómica recesiva. En todas las familias, la tetra-amelia-1 se asoció con malformaciones graves de otras partes del cuerpo, como la cara y la cabeza, anomalías del sistema nervioso, el esqueleto y los genitales. Los pulmones estaban subdesarrollados en muchos casos, lo que dificulta o imposibilita la respiración . Zimmer et al. reportó en 1985 una familia fuertemente endogámica en la que 6 bebés tenían tetra-amelia-1 e hidrocefalia. Describieron en uno de los fetos una ausencia total de hueso pélvico, labio y paladar hendido, arrinia y aplasia de las orejas. También se observó un pulmón izquierdo bilateral, un canal arterial persistente, una imperforación anal. Las pruebas fetales eliminaron el diagnóstico del síndrome de Robert . Otros casos recuperados en la literatura incluyen el de Kosaki et al., en 1996, con un feto de cariotipo 46, XX con tetrafocomelia e hipoplasia pulmonar severa además de anomalías en la cara y la cabeza . Rosenak et al. se describió un caso de tetra-amelia con hipoplasia pulmonar grave en dos fetos de una pareja no consanguínea. Las pruebas fetales descartaron el diagnóstico de síndrome de Robert . Otros dos casos fueron notificados por Zlotogora et al. en 1993. Ambos pacientes fallecieron poco después del nacimiento y los autores sugirieron la existencia de hipoplasia pulmonar. Niemann et al. informó de una familia turca consanguínea en la que 4 de los 8 hermanos sufrían de tetra-amelia. Además de la ausencia de los 4 miembros, los exámenes fetales de 3 fetos revelaron múltiples anomalías: labio leporino y /o palatino, laparosquisis, anomalías pulmonares, hipoplasia de la pelvis, atresia de las choanas, vagina e imperforación anal . Finalmente, en 2005, Krahn et al. se describieron 2 hermanos nacidos de padres consanguíneos que sufrían de tetraamelia e hipoplasia pulmonar grave. Las clavículas y los omóplatos eran normales en el segundo feto. El cariotipo era normal .
El síndrome de Tetra-amelia-1 o TETAMS1 es causado por una mutación homocigota en el gen WNT3 en el cromosoma 17q21 con una herencia autosómica recesiva. El síndrome de tetraamelia-2 (TETAMS2) se caracteriza por extremidades rudimentarias o una ausencia completa de las extremidades, generalmente simétricas, así como agenesia bilateral de los pulmones en algunos casos. También son anomalías habituales del sistema vascular pulmonar y dismorfias que incluyen labio leporino y paladar hendido bilateral, anquiloglosia, hipoplasia mandibular, microrretrognatia y aplasia labioscrotal .
Szenker-Ravi, al estudiar 4 familias de tetra-amelia con agenesia o hipoplasia pulmonar, observó una heterogeneidad fenotípica con anomalías de las extremidades de gravedad variable . La secuenciación del exoma en estas 4 familias ha permitido identificar mutaciones homocigotas truncantes en el gen RSPO2 . El síndrome de tetraamelia-2 es causado por una mutación homocigótica en el gen RSPO2 (610575) localizado en el cromosoma 8q23 .
El fenotipo del primer caso descrito en este artículo corresponde a un síndrome de tetra-amelia-1 debido en particular a la presencia de hidrocefalia, anomalías genitales y nariz rudimentaria. El pecho estrecho y la muerte prematura antes de los 10 minutos de vida sugieren hipoplasia pulmonar grave. Este caso destaca la heterogeneidad fenotípica con coloboma de párpados, hipertelorismo, exoftalmos y apéndices raros.
Consideramos que el segundo caso de nuestro estudio es un síndrome de tetramelia-2 considerando la tetra-amelia simétrica con presencia de muñones en las extremidades superiores. El diagnóstico de la tetra-amelia se debe realizar temprano durante la monitorización por ultrasonido. Por lo tanto, se debe crear conciencia sobre la importancia del monitoreo por ultrasonido y el uso de 3D/4D para mejorar los resultados de los exámenes de detección. El diagnóstico de una masa de pelvis en el ultrasonido emparejado con amelia debe levantar la sospecha de síndrome de defecto de fusión espleno-gonadal de las extremidades.
Además, el examen fetal y las pruebas fetales utilizando las tecnologías en evolución de microarrays cromosómicos y secuenciación de exomas y genomas deben fomentarse en nuestros entornos. Una mejor caracterización de los casos permite asesorar a las parejas y conocer mejor estas anomalías clínicas.
Conclusión
El síndrome de Tetra-amelia es escaso y aún quedan áreas grises. Estos dos casos, en comparación con lo ya descrito en la literatura, ilustran la heterogeneidad fenotípica de la tetraamelia. Dada la rara incidencia de estas anomalías, sería importante crear un registro internacional de anomalías para informar de los casos y establecer un banco de muestras para estudios genéticos extendidos a los padres.
- Wilcox WR, Coulter CP, Schmitz ML (2015) Trastornos por deficiencia congénita de las extremidades. Clin Perinatol 42: 281-300.
- Bermejo SE, Cuevas L, Amar E (2011) Amelia: A multi-center descriptive epidemiologic study in a large dataset from the International Clearinghouse for Birth Defects Surveillance and Research, and overview of the literature. Am J Med Genet C Semin Med Genet 157: 288-304.
- Zlotogora JSM, Shabany YO, Jarallah RY (1993) Síndrome de tetraamelia con hipoplasia pulmonar. Am J Med Genet 47: 570-571.
- Zimmer EZ (1985)Tetra-amelia con múltiples malformaciones en seis fetos masculinos en un pariente. Europ. J Pediat 144: 412-414.
- Rosenak D (1991) Tetraamelia recurrente e hipoplasia pulmonar con múltiples malformaciones en sib. Am J Med Genet 38: 25-28.
- Gershoni BR (1990) ¿Síndrome de Roberts o «Amelia ligada al cromosoma X» ? . Am J Med Genet 37: 569-572.
- Kosaki K, Jones MC, Stayboldt C (1996) Zimmer phocomelia: delineación por análisis de coordenadas principales. Am J Med Genet 66: 55-59.
- La mutación homocigótica WNT3 de Niemann S(2004) causa tetra-amelia en una gran familia consanguínea. Am J Hum Genet 74: 558-563.
- Krahn M (2005) Síndrome de Tetra-amelia y aplasia pulmonar: informe de una nueva familia y exclusión de genes candidatos. Clin Genet 68: 558-560.
- Szenker-RE, Altunoglu U (2018) La inhibición de RSPO2 de RNF43 y ZNRF3 gobierna el desarrollo de las extremidades independientemente de LGR4/5/6. Nature 557: 564-569.