Síndrome de Aicardi-Goutières: Espectro fenotípico y genético en una serie de tres casos | Anales de Pediatría
El síndrome de Aicardi-Goutières (SGA) es una enfermedad hereditaria rara cuya prevalencia exacta se desconoce. Fue descrita por primera vez en 1984 por Jean Aicardi y Francoise Goutières como una encefalopatía progresiva con inicio en los primeros meses de vida caracterizada por linfocitosis del líquido cefalorraquídeo (LCR) y calcificaciones en los ganglios basales.1 Se manifiesta con irritabilidad, retraso psicomotor, espasticidad, distonía, crisis epilépticas, episodios recurrentes de fiebre aséptica y microcefalia. La mortalidad es mayor durante la fase encefalopática, y aunque la enfermedad típicamente se estabiliza después, causa secuelas neurológicas graves. Otros rasgos característicos que pueden aparecer durante su curso son sabañones, síntomas oculares (principalmente glaucoma), afectación cardíaca o trastornos autoinmunes.2 Los interferones tipo I juegan un papel crucial en la patogénesis de los AGS, en la que su expresión se regula al alza, lo que lleva a un aumento de la producción.3 Por esta razón, uno de los hallazgos de laboratorio clásicos en estos pacientes es un nivel elevado de interferón alfa en el LCR, junto con pleocitosis y niveles igualmente elevados de neopterina y biopterina. Actualmente se está investigando la utilidad potencial de evaluar el nivel de expresión de genes estimulados por interferón en sangre periférica como marcador, ya que hay evidencia de que estos niveles permanecen altos más allá de la fase encefalopática («firma de interferón»).3-5 Otra característica clave es la detección de anomalías de neuroimagen, como calcificaciones en los ganglios basales y cambios en la materia blanca (Fig. 1). Hasta la fecha, conocemos 7 genes cuyas mutaciones pueden conducir a una regulación ascendente de la vía del interferón: ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1 e IFIH1. Se han descrito mutaciones heterocigotas para los genes TREX1, ADAR e IFIH1, mientras que las mutaciones reportadas en todos los otros genes han sido homocigotas.2 Las mutaciones en el gen IFIH1 se detectaron más recientemente (2014)4 y, por lo tanto, son las variantes patógenas menos frecuentes, mientras que las mutaciones en los genes RNASEH2B y TREX1 representan la mayor proporción de casos diagnosticados de AGS.
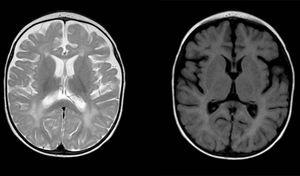
Anomalías de señal difusas y irregulares en materia blanca en ambos hemisferios cerebrales, hiperintensas en imágenes ponderadas en T2. Agrandamiento del espacio subaracnoideo con predominio frontotemporal en ambos hemisferios, con ensanchamiento de la fisura interhemisférica y aumento del tamaño ventricular (en ausencia de aumento de la presión), compatible con atrofia cortical y subcortical.
En las últimas décadas, gracias a los avances en genética que permiten la detección de estas mutaciones específicas, ha surgido evidencia de un amplio espectro fenotípico más allá de la presentación clásica basada en el gen causante. Presentamos los casos de 3 pacientes diagnosticados de SGA en los últimos 8 años con el objetivo de analizar sus características clínicas en relación con el defecto genético subyacente (Tabla 1). En general, las características de presentación del AGS fueron consistentes con las descritas en las series de casos más recientes de la literatura: presentación neonatal (33%), microcefalia (66%), retraso psicomotor (100%), espasticidad (100%), discapacidad intelectual grave (66%) y calcificaciones en TC craneal (66%), aunque solo un paciente tuvo crisis epilépticas.
Características de los pacientes con síndrome de Aicardi-Goutières.
| Caso 1 | Caso 2 | Caso 3 | |
|---|---|---|---|
| la Genética | mutación Homocigótica (p.Ala177Thr) en RNASEH2B gen | mutación Homocigótica (341G>A) en TREX1 gen | mutación Heterocigota (c.992C>G y p.Thr331Arg) en el gen IFIH1 |
| edad Actual | 3 años | 7 años y 4 meses | 12 años y 11 meses |
| Sexo | Varón | Mujer | Macho |
| Origen | Rumania | España | Italia |
| AP | – | Semana 36: restricción del crecimiento intrauterino de la Semana 37: microcefalia, calcificación placentaria |
Paladar hendido |
| Manifestaciones clínicas | |||
| Edad inicial | 10 meses | Nacimiento | 2 años |
| Presentación inicial | Irritabilidad Regresión psicomotora |
Temblores, hipotonía, llanto débil, retraso en el crecimiento | Retraso en la habilidad motora |
| Retraso psicomotor | Sí | Sí | Sí |
| Idioma | 2 sílabas words | No | Yes |
| Microcephaly | Yes | Yes | No |
| Chilblains | No | Yes | Yes |
| Epilepsy | No | Yes | No |
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFCS IV |
| trastorno de Movimiento | No | Sí | No |
| movimientos oculares Anormales | No | No | No |
| discapacidad Visual | No | – | la Miopía |
| el Glaucoma | No | No | No |
| la pérdida de la Audición | – | – | No |
| La afectación cardíaca | No | Leve tricúspide y mitral con regurgitación | No |
| fiebre Recurrente | No | No | No |
| discapacidad Intelectual | Sí | Sí, grave | Sí, leve |
| Otros | – | – | Singleton-Merten síndrome de: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
| CSF | |||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – |
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – |
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – |
| INF alfa (IU/mL) | 25 ( | 75 ( | – |
| Blood | |||
| Interferon signature | – | – | Gene overexpression |
| Neuroimaging | |||
| Calcifications on CT | No | Yes. En ganglios basales y periventriculares | Sí. Calcificaciones simétricas en WM profundo de la región frontal y el núcleo lentiforme |
| Resonancia magnética de la cabeza | Cambios difusos e irregulares en la intensidad de WM de ambos hemisferios cerebrales, hiperintensos en T2. Afectación de la masa muscular subcortical (preservando las fibras U) y de la masa muscular periventricular | Afectación generalizada de la masa muscular con predominio de la masa muscular lobar, incluidas las fibras U subcorticales de los lóbulos frontal, temporal y occipital, bilateral y simétricamente, sin afectación cortical | – |
LCR: líquido cefalorraquídeo; TC: tomografía computarizada; GMFC: Sistema de Clasificación de la Función Motora Gruesa; INF: interferón; RCIU: restricción del crecimiento intrauterino; RMN: imágenes por resonancia magnética; PNP: polineuropatía; WM: madre blanca.
Como se señaló anteriormente, las mutaciones homocigotas en el gen RNASEH2B son las variantes más frecuentes que causan AGS y su expresión fenotípica generalmente se ajusta más a la presentación clásica.4 Este fue el caso de la paciente de nuestro estudio portadora de dicha mutación, que comenzó a los 10 meses con irritabilidad y retraso psicomotor y con hallazgos característicos de neuroimagen y LCR.
El veinte por ciento de los casos de AGS pueden tener una presentación neonatal, con el inicio de la enfermedad en el útero.5 Mutaciones en cualquiera de los 7 genes mencionados anteriormente pueden conducir a este fenotipo, pero esta presentación temprana se asocia con mayor frecuencia con el gen TREX.4,5 La presentación inicial de esta forma es similar a la de una infección por TORCH, con hepatoesplenomegalia, hipertransaminasemia, trombocitopenia y manifestaciones neurológicas que incluyen irritabilidad extrema, trastornos del movimiento y convulsiones epilépticas.5 Estos pacientes tienen un curso de enfermedad más grave y tienen un mayor riesgo de muerte. El paciente de nuestra muestra que presentó una variante de este tipo tenía una presentación neonatal y actualmente tiene la forma de enfermedad más grave de las 3.
Las mutaciones en el gen ADAR1 y especialmente en el gen IFIH1 se asocian con un inicio tardío de los síntomas, después de 1 año de vida con desarrollo psicomotor normal.5 En algunos de estos casos, el síndrome tiene un curso benigno con preservación relativa del lenguaje y las habilidades motoras. Nuestro paciente con una mutación en el gen IFIH1 fue un caso singular en el que también padecía síndrome de Singleton-Merten, una enfermedad rara también causada por una mutación en el gen IFIH1 y caracterizada por displasia dental, calcificaciones aórticas y osteoporosis.6
Nuestro objetivo es subrayar la significativa variabilidad fenotípica del SGA y su asociación con mutaciones específicas con el fin de fomentar la consideración de este diagnóstico en casos con presentaciones que se desvían de la forma clásica de la enfermedad y de aportar información adicional sobre el curso de la enfermedad y los resultados en estos pacientes.