Aicardi-Goutièresin oireyhtymä: fenotyyppi ja geneettinen kirjo kolmen tapauksen sarjassa | Anales de Pediatría
Aicardi-Goutièresin oireyhtymä (AGS) on harvinainen perinnöllinen sairaus, jonka tarkkaa esiintyvyyttä ei tunneta. Jean Aicardi ja Francoise Goutières kuvasivat sen ensimmäisen kerran vuonna 1984 eteneväksi enkefalopatiaksi, joka puhkeaa ensimmäisten elinkuukausien aikana ja jolle on ominaista aivo-selkäydinnesteen lymfosytoosi ja tyvitumakkeiden kalkkeutumiset.1 se ilmenee ärtyneisyytenä, psykomotorisena jälkeenjääneisyytenä, spastisuutena, dystoniana, epileptisinä kohtauksina, toistuvina aseptisen kuumeen ja mikrokefalian kohtauksina. Kuolleisuus on suurempi enkefalopaattisen vaiheen aikana, ja vaikka tauti tyypillisesti stabiloituu jälkeenpäin, se aiheuttaa vakavia neurologisia jälkiseurauksia. Muita tyypillisiä piirteitä, jotka voivat näkyä sen aikana, ovat kylmät, silmäoireet (pääasiassa glaukooma), sydänoireet tai autoimmuunisairaudet.2 tyypin I interferoneilla on ratkaiseva merkitys AGS: n patogeneesissä, jossa niiden ilmentymistä säädellään ja tuotanto lisääntyy.3 tästä syystä yksi klassisista laboratoriolöydöksistä näillä potilailla on kohonnut interferoni alfan pitoisuus aivo-selkäydinnesteessä sekä plesosytoosi ja yhtä suuret neopteriini-ja biopteriinipitoisuudet. Parhaillaan tutkitaan, onko interferonilla stimuloitujen geenien ilmentymisen markkeritasoa perifeerisessä veressä mahdollista hyötyä arvioitaessa, koska on näyttöä siitä, että nämä pitoisuudet pysyvät korkeina yli enkefalopaattisen vaiheen (”interferonijälki”).3-5 toinen keskeinen piirre on neuroimaging poikkeavuuksia kuten kalkkeutumista tyvitumakkeessa ja muutoksia valkoisen aineen (Fig. 1). Tähän mennessä tiedämme 7 geeniä, joiden mutaatiot voivat johtaa interferonireitin säätelyyn: ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1 ja IFIH1. Heterotsygoottisia mutaatioita on kuvattu TREX1 -, ADAR-ja IFIH1-geeneille, kun taas kaikissa muissa geeneissä raportoidut mutaatiot ovat olleet homotsygootteja.2 mutaatioita IFIH1-geenissä havaittiin viimeksi (2014) 4 ja ovat siksi vähiten toistuvia patogeenisiä variantteja, kun taas mutaatiot RNASEH2B-ja TREX1-geeneissä aiheuttavat suurimman osan diagnosoiduista AGS-tapauksista.
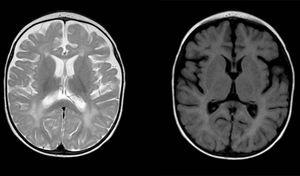
diffuusi ja hajanainen signaalipoikkeavuus valkoisessa aineessa molemmilla aivopuoliskoilla, hyperintensiteetti T2-painotetuissa kuvissa. Laajentunut subaraknoidinen tila frontotemporaalinen hallitsevuus molemmilla pallonpuoliskoilla, laajentamalla interhemispheric halkeama ja lisääntynyt kammion koko (ilman lisääntynyt paine), yhteensopiva aivokuoren ja subkortiaalinen atrofia.
viime vuosikymmeninä genetiikan edistymisen ansiosta, joka mahdollistaa näiden erityisten mutaatioiden havaitsemisen, on ilmaantunut todisteita laajasta fenotyyppisestä kirjosta, joka ylittää klassisen esityksen, joka perustuu aiheuttavaan geeniin. Esitämme tapaukset, joissa 3 potilasta on saanut AGS-diagnoosin viimeisten 8 vuoden aikana tarkoituksena analysoida niiden kliinisiä ominaisuuksia suhteessa taustalla olevaan geenivirheeseen (Taulukko 1). AGS: n ominaisuudet olivat yleisesti ottaen yhdenmukaiset kirjallisuuden viimeisimmässä tapaussarjassa kuvattujen piirteiden kanssa: vastasyntyneen esiintyminen (33%), mikrokefalia (66%), psykomotorinen jälkeenjääneisyys (100%), spastisuus (100%), vaikea kehitysvammaisuus (66%) ja kallon TT: n kalkkeutuminen (66%), vaikka vain yhdellä potilaalla oli epileptisiä kohtauksia.
Aicardi-Goutièresin oireyhtymää sairastavien potilaiden ominaisuudet.
| Case 1 | Case 2 | Case 3 | |
|---|---|---|---|
| genetiikka | homotsygoottinen mutaatio (P.Ala177Thr) RNASEH2B-geenissä | homotsygoottinen mutaatio (341g>a) TREX1-geenissä | heterotsygoottinen mutaatio (c.992C>G ja p.Thr331Arg) IFIH1-geenissä |
| nykyinen ikä | 3 vuotta | 7 vuotta ja 4 kuukautta | 12 vuotta ja 11 kuukautta |
| sukupuoli | mies | nainen | mies |
| alkuperä | Romania | Espanja | Italia |
| AP | – | viikko 36: kohdunsisäisen kasvun rajoitus viikko 37: mikrokefalia, istukan kalkkeutuminen |
suulakihalkio |
| kliiniset oireet | |||
| Ikä alkamisvaiheessa | 10 kuukautta | syntymä | 2 vuotta |
| Alkuesitys | ärtyneisyys psykomotorinen regressio |
vapina, hypotonia, heikko itku, kasvuhäiriö | motorisen taidon viive |
| psykomotorinen jälkeenjääneisyys | Kyllä | Kyllä | Kyllä |
| kieli | 2-tavuinen words | No | Yes |
| Microcephaly | Yes | Yes | No |
| Chilblains | No | Yes | Yes |
| Epilepsy | No | Yes | No |
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFC-yhdisteet IV |
| liikehäiriö | Ei | Kyllä | Ei |
| epänormaalit silmien liikkeet | Ei | Ei | Ei |
| näköhäiriöt | Ei | – | likinäköisyys |
| glaukooma | Ei | Ei | Ei |
| kuulon heikkeneminen | – | – | Ei |
| sydänoireet | Ei | lievä kolmiliuskaisuus ja mitraalinen regurgitaatio | Ei |
| uusiutuva kuume | Ei | Ei | Ei |
| kehitysvammaisuus | Kyllä | Kyllä, vakava | Kyllä, lievä |
| muuta | – | – | Singleton-Mertenin oireyhtymä: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
| CSF | |||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – |
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – |
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – |
| INF alfa (IU/mL) | 25 ( | 75 ( | – |
| Blood | |||
| Interferon signature | – | – | Gene overexpression |
| Neuroimaging | |||
| Calcifications on CT | No | Yes. Tyvitumakkeessa ja periventrikulaarisessa gangliassa | Kyllä. Symmetriset kalkkeutumat otsan alueen ja tunnistavan tuman syvässä WM: ssä |
| pään MK | diffuusi ja hajanainen WM-voimakkuus molemmissa aivopuoliskoissa, hyperintensiteetti T2: ssa. Subkortiaalisen WM: n (u-kuituja säästäen) ja periventrikulaarisen WM | yleinen WM: n sekaantuminen, jossa suurin osa lobar WM: stä, mukaan lukien otsa -, ohimo-ja takaraivolohkojen subkortiaaliset U-kuidut, molemminpuolisesti ja symmetrisesti, ilman kortikaalista osuutta | – |
aivo-selkäydinneste; CT, tietokonetomografia; GMFC-yhdisteet, Karkeamotorisen toiminnan luokitusjärjestelmä; inf, interferoni; IUGR, kohdunsisäisen kasvun rajoitus; MK, magneettikuvaus; PNP, polyneuropatia; WM, white mater.
kuten aiemmin todettiin, homotsygoottiset mutaatiot RNASEH2B-geenissä ovat yleisimpiä variantteja, jotka aiheuttavat AGS: ää ja niiden fenotyyppinen ilmentyminen noudattaa yleensä eniten klassista esitystapaa.4 Näin kävi tutkimuksemme potilaalle, jolla oli tällainen mutaatio, joka oli alkanut 10 kuukauden iässä ärtyneisyydellä ja psykomotorisella jälkeenjääneisyydellä sekä tyypillisillä neuroimaging-ja aivo-SELKÄYDINNÄYTTEILLÄ.
kaksikymmentä prosenttia AGS-tapauksista saattaa esiintyä vastasyntyneillä, jolloin tauti alkaa kohdussa.5 mutaatiot missä tahansa 7 edellä geeneissä voivat johtaa tähän fenotyyppiin, mutta tämä varhainen esiintyminen liittyy useimmin TREX-geeniin.4, 5 tämä muoto on aluksi samanlainen kuin SOIHTUINFEKTIO, johon liittyy hepatosplenomegaliaa, hypertransaminasemiaa, trombosytopeniaa ja neurologisia oireita, kuten äärimmäistä ärtyneisyyttä, liikehäiriöitä ja epileptisiä kohtauksia.5 Näiden potilaiden sairaus on vakavampi ja heillä on suurempi riski kuolla. Näytteemme potilaalla, joka esitti tällaisen variantin, oli vastasyntyneen esitys ja tällä hetkellä 3: n vakavin sairaus.
Adar1-geenin ja erityisesti IFIH1-geenin mutaatiot liittyvät oireiden myöhäiseen alkamiseen, 1-elinvuoden jälkeen normaaliin psykomotoriseen kehitykseen.5 joissakin näistä tapauksista oireyhtymä on hyvänlaatuinen kurssi suhteellinen säilyttäminen kielen ja motorisia taitoja. Potilaamme, jolla oli mutaatio IFIH1-geenissä, oli ainutlaatuinen tapaus siinä mielessä, että hänellä oli myös Singleton-Mertenin oireyhtymä, harvinainen sairaus, jonka aiheutti myös mutaatio IFIH1-geenissä ja jolle on ominaista hampaiden dysplasia, aortan kalkkeutuminen ja osteoporoosi.6
tavoitteenamme on korostaa AGS: n merkittävää fenotyyppistä vaihtelua ja sen yhteyttä tiettyihin mutaatioihin sekä kannustaa tämän diagnoosin tarkasteluun tapauksissa, joissa esitystavat poikkeavat taudin klassisesta muodosta, että antaa lisätietoa taudin kulusta ja tuloksista näillä potilailla.