jaksolliset paralyysit
taulukossa 1 esitetty primaaristen jaksollisten paralyysien kliinisesti hyödyllinen luokittelu sisältää hypokaleemisia, hyperkaleemisia ja paramyotonisia muotoja.
Taulukko 1. Primaarinen periodinen halvaus (muokattu Jurkat-Rottista ja Lehmann-Hornista) (avoin pöytä uudessa ikkunassa)
|
tauti |
Gene |
proteiini |
perintö |
mutaatio |
|
HyperPP |
SCN4A |
Nav1.4 |
dominoiva |
hyvä on. |
|
NormoPP |
hieno (ω-pore) |
|||
|
Paramyotoniacongenita |
hyvä on. |
|||
|
HypoPP tyyppi II |
hieno (ω-pore) |
|||
|
HypoPP-Järjestys |
CACNA1S |
Cav1.1 |
dominoiva |
voitto (ω-pore) |
|
ThyrotoxicPP |
KCNJ18 |
Kir2.18 |
dominoiva |
tappio |
|
Andersen-Tawilin oireyhtymä |
KCNJ2 |
Kir2.1 |
dominoiva |
tappio |
flaccid-heikkouden fysiologinen perusta on lihaskalvon (eli sarkolemman) epätäsmällisyys. Seerumin kaliumpitoisuuden muuttuminen ei ole primaarisen PP: n pääasiallinen vika; kaliumin metabolian muuttuminen johtuu PP: stä. Primaarisessa ja tyrotoksisessa PP: ssä veltto halvaus tapahtuu suhteellisen pienillä seerumin kaliumpitoisuuden muutoksilla, kun taas sekundaarisessa PP: ssä seerumin kaliumpitoisuudet ovat selvästi epänormaaleja.
mikään yksittäinen mekanismi ei ole vastuussa tästä häiriöryhmästä. Ne ovat siis heterogeenisiä, mutta niillä on joitakin yhteisiä piirteitä. Heikkous on yleensä yleistynyt, mutta se voi olla paikallinen. Kallon lihaksisto ja hengityslihakset yleensä säästyvät. Venytysrefleksit ovat joko poissa tai heikentyneet hyökkäysten aikana. Lihassyyt ovat sähköistämättömiä hyökkäysten aikana. Lihasvoima on normaalia hyökkäysten välillä, mutta muutaman vuoden kuluttua tietyntyyppisiin PP: hen (erityisesti primaariseen PP: hen) kehittyy jonkinasteinen kiinteä heikkous. Kaikki primaarisen PP: n muodot (Becker myotonia congenitaa lukuun ottamatta ) ovat joko autosomaalisesti dominoivia periytyviä tai sporadisia (jotka johtuvat todennäköisimmin pistemutaatioista).
Jänniteherkät ionikanavat säätelevät tarkasti toimintapotentiaalien muodostumista (solukalvojen jännitteen lyhyet ja palautuvat muutokset). Nämä ovat selektiivisesti ja vaihtelevasti läpäiseviä ionikanavia. Energiasta riippuvaiset ionikuljettajat ylläpitävät konsentraatiogradientteja. Toimintapotentiaalien syntymisen aikana natriumionit liikkuvat kalvon poikki jännitteellä varustettuja ionikanavia pitkin. Lepäävä lihaskuitukalvo polarisoituu pääasiassa kloridin liikkeestä kloridikanavien läpi ja repolarisoituu kaliumin liikkeestä. Natrium -, kloridi-ja kalsiumkanavat ryhmänä liittyvät myotoniaan ja PP: hen. Natrium -, kalsium-ja kaliumkanavien toiminnalliset alayksiköt ovat homologisia. Natriumkanavapatiat ymmärretään paremmin kuin kalsium-tai kloridikanavapatiat. Kaikki familiaalisen PP: n muodot osoittavat lopullisen mekanistisen reitin, johon liittyy poikkeava depolarisaatio, inaktivoivat natriumkanavat ja lihassyyn inexcitability.
tässä artikkelissa käsitellään ensisijaisesti natrium -, kalsium-ja kaliumkanavapatioita sekä PP: n toissijaisia muotoja. Kloridikanavapatiat eivät liity episodiseen heikkouteen, ja niitä käsitellään yksityiskohtaisemmin myotonisia häiriöitä käsittelevissä artikkeleissa.
Yhteenveto kanavan toimintahäiriöistä eri PP-tyypeissä
HyperPP-nopean kanavan inaktivaation myötä mutaatiot sijaitsevat yleensä transmembraanisegmenttien sisäosissa tai solunsisäisissä silmukoissa, jotka vaikuttavat nopean inaktivoituvan hiukkasen telakointipaikkoihin, mikä heikentää nopean kanavan inaktivaatiota ja johtaa pysyvään Na+ – virtaan.
Hypopp-hyperpolarisaatioaktivoidun kationivuodon torjuessa K+ – oikaisevaa virtaa mutaatiot aiheuttavat uloimman arginiini-tai lysiinisubstituution.
NormoPP depolarisaatioaktivoidulla kationivuodolla mutaatiot ovat domeeni II: n jännitesensorin syvemmissä paikoissa kodonissa R675.
ionikanavan toimintahäiriöt kompensoidaan yleensä hyvin normaalilla eksitaatiolla, ja lisälaukaisimet ovat usein välttämättömiä, jotta lihas ei hajoaisi, koska kalvot depolarisoituvat jatkuvasti.
glukoosin ja kaliumin saanti vaikuttaa näissä häiriöissä päinvastaisesti. HyperPP: ssä kaliumin saanti laukaisee hyökkäyksen, kun taas glukoosi amelioroi sitä. Sen sijaan glukoosi aiheuttaa hypokaleemisia hyökkäyksiä ja kalium on hyökkäyksen hoito.
huomaa alla oleva kuva.
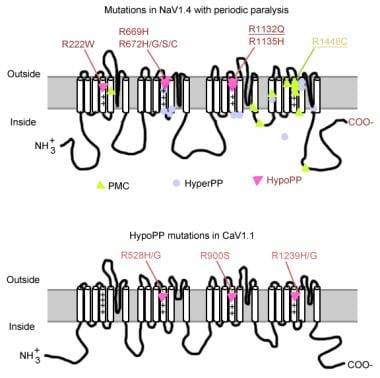 mutaatiot periodisessa halvauksessa.
mutaatiot periodisessa halvauksessa. Lihasnatriumkanavageeni
natriumkanavassa on alfa-alayksikkö ja beeta-alayksikkö. Natriumkanavan alfa-alayksikkö on 260 kd: n glykoproteiini, joka koostuu noin 1800-2000 aminohaposta. Tämä kanava on hyvin säilynyt evolutionaarisesti drosophilasta ihmiseksi. Siinä on 4 homologista domeenia (I-IV), jotka taittuvat muodostaen keskushuokoset, joissa kussakin on 225-325 aminohappoa. Kukin domeeni koostuu 6 hydrofobisesta segmentistä (S1-S6), jotka halkovat solukalvoa. Kanavan tärkeimmät toiminnot ovat jänniteherkkä gatointi, inaktivointi ja ioniselektiivisyys. Solunulkoinen silmukka S5: n ja S6: n välillä dippaa plasmakalvoon ja osallistuu huokosien muodostumiseen. S4-segmentti sisältää positiivisesti varautuneita aminohappoja joka kolmannessa asennossa ja toimii jänniteanturina. Depolarisaation aikana voi tapahtua konformaatiomuutoksia, jotka johtavat kanavan aktivoitumiseen ja inaktivoitumiseen. Solusilmukka verkkotunnuksen III-S6 ja verkkotunnuksen IV-S1 välillä toimii inaktivoivana porttina.
natriumkanavassa on 2 porttia (aktivaatio ja inaktivaatio) ja niitä voi olla 3 tilassa. Levossa kalvon polarisoituessa aktivointiportti suljetaan ja inaktivointiportti avataan. Depolarisaatiolla aktivaatioportti avautuu, jolloin natriumionit pääsevät kulkemaan ionikanavan läpi ja altistavat myös telakointipaikan inaktivaatioportille. Depolarisaation jatkuessa inaktivaatioportti sulkeutuu estäen natriumin pääsyn soluun ja aiheuttaen kanavan nopean inaktivaation tilaan. Tämä kanavan inaktivointi mahdollistaa kalvon repolarisoitumisen, jolloin aktivointiportti suljetaan ja inaktivointiportti avataan takaisin lepotilaan. Nisäkkäiden luustolihaksissa tapahtuu kaksi inaktivaatioprosessia: nopea inaktivaatio tarkoittaa toimintakyvyn loppumista ja vaikuttaa millisekunnin aikajänteellä. Hidas inaktivointi kestää sekunneista minuutteihin ja voi säädellä eksitoituvien natriumkanavien populaatiota.
Natriumkanavamutaatiot, jotka häiritsevät nopeaa ja hidasta inaktivaatiota, liittyvät yleensä HyperPP: n fenotyyppiin ja myotoniaan, jossa hitaasti tai nopeasti inaktivaatiota lisäävinä mutaatioina natriumkanavan toiminnan menetys aiheuttaa HypoPP: n.
natriumkanavageenin (SCN4A) mutaatioilla on useita yleisiä piirteitä. Suurin osa mutaatioista on ”inaktivoivassa” linkkerissä toistojen III ja IV välillä, toistojen IV ”jännitteen tunnistavassa” segmentissä S4 tai sisäkalvossa, jossa ne voisivat heikentää inaktivaatioportin telakointipaikkaa. Kliininen fenotyyppi eroaa spesifisten aminohapposubstituutioiden perusteella, ja vaikka hyperkaleemisten PP: n, paramyotonia congenitan (PC) ja kaliumin pahenemista pahentavien myotonioiden (Pam) välillä saattaa esiintyä jonkin verran päällekkäisyyttä, nämä kolme fenotyyppiä ovat yleensä erillisiä (kuten alla on kuvattu). Lähes kaikki mutanttikanavat ovat heikentäneet natriumvirran nopeaa inaktivoitumista. Useimmat potilaat ovat herkkiä systeemiselle kaliumille tai kylmälle lämpötilalle.
on olemassa kaksi kanavapopulaatiota, mutantti-ja villityyppisiä; heikentynyt nopea inaktivaatio johtaa mutanttilihaskuitukalvojen pitkittyneeseen depolarisaatioon ja voi selittää näiden häiriöiden 2 kardinaalioiretta, myotonian ja heikkouden. Hyperkaleemisessa PP: ssä tapahtuu mutanttikanavan gatingissa toiminnan vahvistuminen, mikä johtaa lisääntyneeseen natriumvirtaan, joka depolarisoi sairasta lihasta liikaa. Myofiber-kalvon lievä depolarisaatio (5-10 mV), joka voi johtua solunulkoisista kaliumpitoisuuksista, johtaa mutanttikanavien säilymiseen noninaktivoituneessa tilassa. Jatkuva sisäänpäin suuntautuva natriumvirta aiheuttaa villin tyypin natriumkanavien toistuvaa laukeamista, mikä koetaan jäykkyytenä (eli myotoniana).
jos ilmenee vakavampi depolarisaatio (20-30 mV), sekä normaalit että epänormaalit kanavat ovat inaktivoituneet aiheuttaen heikotusta tai halvaantumista. Näin ollen hienovaraiset erot kalvon depolarisaation vakavuudessa voivat tehdä eron myotonian ja halvauksen välillä. Lämpötilaherkkyys on PC: n tunnusmerkki. Kylmyys pahentaa myotoniaa ja aiheuttaa heikkoutta. Tähän tilaan liittyy useita mutaatioita, joista 3 on samassa kohdassa (1448) S4-segmentissä. Nämä mutaatiot korvaavat arginiinin muilla aminohapoilla ja neutraloivat tämän hyvin säilyneen S4-positiivisen varauksen. Näiden jäämien mutaatiot ovat PC: n yleisin syy. Joitakin mahdollisia mekanismeja vastuussa lämpötilan herkkyys ovat seuraavat:
-
lämpötila voi vaikuttaa mutanttikanavan konformaatiomuutokseen eri tavoin.
-
matalammat lämpötilat saattavat vakauttaa mutanttikanavat epänormaaliin tilaan.
-
mutaatiot voivat muuttaa kanavan herkkyyttä muille soluprosesseille, kuten fosforylaatiolle tai toisille viestimille.
useimmat hyperkaleemiset PP-tapaukset johtuvat 2 mutaatiosta scn4a: ssa, T704M: ssä ja M1592V: ssä. Mutaatiot natrium-kanavan, erityisesti jäämien 1448 ja 1313, ovat vastuussa paramyotonia congenita. Pieni osa hypokalemia, määräajoin halvaus tapauksissa liittyy mutaatioita kodonien 669 ja 672 (HypoPP2). Vuonna HypoPP2, natrium kanava mutaatioita parantaa inaktivaatio tuottaa nettotappio toiminto vika.
Normokaleeminen PP muistuttaa sekä HyperPP: tä (kaliumherkkyys) että HypoPP: ia (hyökkäysten kesto) ja sen aiheuttavat SCN4A-mutaatiot jännitesensori dii: n syvemmässä paikassa kodonissa 675. R675-mutaatiot eroavat HypoPP-mutaatioista siinä, että nämä mutaatiot johtavat depolarisaatioaktivoituun poreen, joka tuottaa ω-virran käänteisellä jänniteriippuvuudella, koska tämä sivusto altistuu solunulkoisille paikoille voimakkaammalla depolarisaatiolla.
Kalsiumkanavageeni
kalsiumkanavageeni (CACNL1A3) on 5 alayksikön (alfa-1, alfa-2, beeta, gamma ja delta) muodostama kompleksi. Luurankolihaksen DIHYDROPYRIDIINIRESEPTORI (DHP) sijaitsee pääasiassa poikittaisessa tubuluskalvossa. Alfa-1-alayksikössä on SITOUTUMISPAIKKOJA DHP-lääkkeille ja se johtaa hidasta L-tyypin kalsiumvirtaa. Se osallistuu myös excitation-contraction (EC) – kytkentään ja toimii jännitesensorina kytkemällä sen sarkoplasmisen retikulumin ryanodiinireseptoriin (eli kalsiumin vapautuskanavaan). Kalvopotentiaalin muutokset liittyvät solunsisäiseen kalsiumin vapautumiseen, mikä mahdollistaa EC-kytkennän. DHP-reseptorin/kalsiumkanavan alfa-1-alayksikön pistemutaatiot aiheuttavat hypokaleemista PP: tä (HypoPP1). Kaksi cacna1s-geenin mutaatiota, R528H ja R1239H, ovat vastuussa useimmista hypokaleemisista PP-tapauksista.
sairauden fysiologista perustaa ei edelleenkään ymmärretä, vaan se johtuu todennäköisemmin eksitaation epäonnistumisesta kuin EC-kytkennän pettämisestä. Hypokalemian aiheuttama depolarisaatio voi kuitenkin vähentää kalsiumin vapautumista, mikä vaikuttaa kanavan jännitteen säätelyyn suoraan tai välillisesti natriumkanavan inaktivoinnin kautta. Insuliini ja adrenaliini voivat vaikuttaa samalla tavalla. Kalsiumkanavageenin mutaatioilla on joitakin yhtäläisyyksiä SCN4A-mutaatioihin. Mutaatiot muokkaavat kanavan inaktivaatiota, mutta eivät jänniteriippuvaista aktivaatiota. Myotube-viljelmien tallenteet sairastuneilta potilailta osoittivat DHP-herkän L-tyypin kalsiumvirran vähentyneen 30%. Kanavat inaktivoituvat matalissa kalvopotentiaaleissa.
kalsiumkanavan mutaatiot aiheuttavat toiminnan heikkenemistä, joka ilmenee alentuneena virrantiheytenä ja hitaampana inaktivaationa. Sitä, miten tämä inaktivaatio liittyy hypokalemian aiheuttamiin hyökkäyksiin, ei ymmärretä. Ainakin R528H-mutaatiossa tapahtuu mahdollinen sekundaarinen kanavointisairaus, joka on sidottu muuttuneesta kalsiumin homeostaasista johtuvan ATP-herkän kaliumvirran pienenemiseen. Cacnl1a3-mutaatioihin liittyvät pienemmät virtaukset voivat hieman muuttaa solunsisäistä kalsiumhomeostaasia, mikä voi vaikuttaa k+-kanavien ominaisuuksiin ja ilmentymiseen, erityisesti sisäänpäin tasasuuntaajakanavien luokkaan kuuluvaan KATP: hen (ATP-herkkä kaliumkanava). Insuliini vaikuttaa myös HypoPP: iin vähentämällä tätä sisäänpäin suuntautuvaa tasasuuntaajan K+ – virtaa.
Jännitesensorin varaushäviö selittää suurimman osan HypoPP-tapauksista. Natrium-ja kalsiumkanavilla on homologisia huokosia muodostavia alfa-alayksiköitä. Cacnl1a3: n ja SCN4A: n pistemutaatiot vaikuttavat Argentiinan jäämiin näiden kanavien S4-jännitesensoreissa. Arginiinimutaatiot S4-segmenteissä aiheuttavat 90% HypoPP-tapauksista.
Jännitesensorin varaushäviö selittää suurimman osan HypoPP-tapauksista. Natrium-ja kalsiumkanavilla on homologisia huokosia muodostavia α-alayksiköitä. Lähes kaikki cav1.1: n (HypoPP-1) ja Nav1.4: n (HypoPP-2) mutaatiot neutraloivat positiivisesti varautuneen aminohapon yhdessä jännitesensorien uloimmista arginiineista tai lysiineistä. Nav1.4 mutaatiota sijoittuu yleisimmin I, II ja III toistojen jännitesensoreihin aiheuttaen kationivuodon.
uloimman arginiinin korvaaminen pienemmällä aminohapolla, kuten glysiinillä, avaa johtavan reitin hyperpolarisoituneessa potentiaalissa, mikä johtaa sisäänpäin kääntyvään kationivirtaan (kationivuoto tai ω-virta, joka erotetaan (ω–) Ionia johtavalla huokosella, on monovalenttien kationien hyperpolarisaatioaktivoitu virta S4-poreen kautta, joka estää oikaisevat K+ – virrat) depolarisoivaa tai epävakauttavaa lepopotentiaalia.
S4-segmentti liikkuu ulospäin depolarisaation aikana sulkien johtavan reitin. Lihassyyt, joilla on vakavia jännitesensorimutaatioita, depolarisoituvat paitsi hypokalemian aikana myös kaliumpitoisuuksissa normaalialueella, mikä selittää interictal – ja pysyvän heikkouden. Vaikeaa myopatiaa, johon liittyy lihaskudoksen rasvoittumista, esiintyy yleisesti potilailla, joilla on Cav1.1 R1239H (DIV-mutaatiot).
glukokortikosteroidit aiheuttavat Hypoppia stimuloimalla insuliinin ja amyliinin välittämää Na+ K+ Atpaasia.
Kaliumkanavageeni
sisäänpäin oikaiseminen on tärkeä kir-kanavien ominaisuus. Oikaisuun liittyy jänniteriippuvainen johtuminen-huokosten tukos polyamiineilla ja Mg++: lla depolarisaation aikana, ja tämä tukos poistetaan potentiaaligradientin aikana hyperpolarisaation aikana. Kaliumkanavamutaatioita esiintyy Andersen-Tawilin oireyhtymässä ja tyrotoksisessa PP: ssä.
Andersen-Tawilin oireyhtymälle on ominaista dysmorfisten piirteiden, jaksollisen halvauksen ja sydämen rytmihäiriöiden kolmijako. Oireyhtymään liittyy kcnj2-geenin mutaatioita. Kcnj2-geeni koodaa sisäänpäin kääntyvää kaliumkanavaa Kir2.1. Kaliumkanavan mutaatioiden kcne3: ssa on raportoitu aiheuttavan hypokaleemista PP: tä, mutta tätä ei ole todistettu.
Kir2.6: n mutaatiot aiheuttavat herkkyyttä tyrotoksiselle PP: lle. Tyrotoksisessa PP: ssä havaittu episodinen heikkous on samankaltainen kuin Hypoppin ja Andersen-Tawilin oireyhtymässä. Tämä häiriö on yleisin aasialaisilla ja latinalaisamerikkalaisilla miehillä. Thyrotoxic PP on geneettinen sairaus, jonka tyrotoxicosis on paljastanut. Kir2. 6 ilmenee pääasiassa luurankolihaksissa. Trijodityroniini parantaa KCNJ18-transkriptiota, joka voi ajaa kir2.6: n tehostettua ilmentymistä. PKC aktivoituu tyrotoksikoosin aikana, koska pip2: n liikevaihto kasvaa ja Kir-kanavat ovat suorassa vuorovaikutuksessa PIP2: n kanssa normaalin gatingin aikana. Andersen-Tawilin oireyhtymässä PIP2-affiniteetti on vähentynyt. Tyrotoksisessa PP: ssä mikään mutaatioista ei muuta Kir2.6: n oikaisua.