Spinal Muscular Atrofy
Causes / Inheritance
What causes spinal muscular atrofy (SMA)?
SMA: lle on tyypillistä motoneuronien eli selkäytimen hermosolujen häviäminen. Se luokitellaan motoneuronitaudiksi.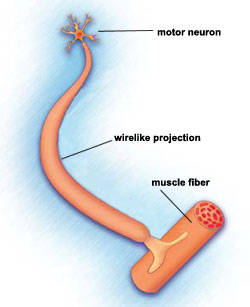
mitkä ovat SMA: n geneettiset syyt?
yleisin SMA: n muoto (tyypit 1-4) johtuu kromosomissa 5 olevan SMN1-geenin virheestä (mutaatiosta). (Ihmisillä on kaksi SMN1-geeniä-yksi jokaisessa kromosomissa 5). 94%: ssa kaikista SMA-tapauksista tämä mutaatio sisältää ekson 7-nimisen segmentin deleetion. Tämä alue sijaitsee kromosomin 5 pitkässä varressa 5q13. 2 alueella (kromosomeilla on kaksi ”haaraa”: lyhyt, joka tunnistetaan p-kirjaimella, ja pitkä, joka tunnistetaan q-kirjaimella).
mutaatio SMN1-geenissä johtaa SMN-nimisen motoneuroniproteiinin puutokseen, joka tulee sanoista ” survival of motor neuron.”Nimensä mukaisesti tämä proteiini vastaa normaalin motoneuronitoiminnan kannalta välttämättömästä geeniekspressiosta.
harvinaisempana mutaatio X-kromosomigeenissä nimeltä UBE1 aiheuttaa X-linkitetyn SMA: n. UBE1-geeni sisältää ohjeet ubikitiiniä aktivoivalle entsyymille 1, joka normaalisti auttaa kiinnittämään molekyylitunnisteen proteiineihin merkitäkseen ne tuhottaviksi.
kromosomissa 14 olevan sytoplasmaisen dynein 1-raskasketjun 1 (DYNC1H1) geenin puutteiden on todettu johtavan toiseen harvinaiseen SMA-muotoon, jota kutsutaan SMA-LEDIKSI.
mikä aiheuttaa SMA: ssa havaitun oireiden vaikeusasteen suuren vaihtelun?
normaalisti SMN1-geenit tuottavat täyspitkää ja täysin toimivaa SMN-proteiinia. Mutta kun SMN1-geenissä on mutaatioita, kuten SMA: n kromosomiin 5 liittyvässä muodossa, SMN-proteiinia ei synny riittävästi.
kromosomissa 5 oleva naapurigeeni, nimeltään SMN2, tuottaa myös SMN-proteiinia. Suurin osa smn2-geenien kuljettamista ohjeista valmistetusta proteiinista ei ole toiminnallista, mutta pieni osa, noin 10-15 prosenttia on toimivaa.
ihmisillä voi olla useita kopioita SMN2-geenistä. Normaalisti määrä vaihtelee nollan ja kahdeksan kappaleen välillä. SMA: n kromosomiin 5 liittyvässä muodossa mitä enemmän smn2-geeniä ihmisellä on, sitä toimivampaa SMN-proteiinia on saatavilla. Tämän seurauksena taudin kulku on todennäköisesti sitä lievempi. Ottaa kolme tai useampia kopioita SMN2 geeni liittyy vähemmän vakava sairaus ilmentymä.
geenitestillä voidaan kertoa, kuinka monta SMN2-geeniä ihmisellä on, ja karkeasti ennustaa SMA: n todennäköisen kulun.
SMA: n vaikeusaste voi myös riippua taudin muuntelijoista, jotka eivät aiheuta sairautta, mutta voivat vaikuttaa (muuttaa) taudin puhkeamiseen ja vaikeusasteeseen vaikuttamalla erilaisiin biologisiin reitteihin. Sekä plastin 3-proteiinin että ZPR1-proteiinin tasot on tunnistettu SMN: ään liittyvän SMA: n modifioijoiksi ja niistä voi tulla terapeuttisia kohteita. Lisäksi näiden proteiinipitoisuuksien testaus voisi auttaa ennustamaan taudin vakavuutta, ja näiden proteiinien toiminnan ymmärtäminen voisi tuoda uutta valoa tautiprosesseihin.

geneettinen informaatio siirtyy varastointimuodostaan DNA: na RNA: ksi kutsuttuun ohjesarjaan, josta proteiinimolekyylit valmistetaan. Suurin osa SMN1-geenin RNA-ohjeista käskee solua tekemään täyspitkää SMN-proteiinia. Suurin osa SMN2-geenin ohjeista käskee solua tekemään lyhyen SMN-proteiinin.
mikä on SMA: n perintökuvio?
kromosomiin 5 liittyvä SMA (tyypit 1-4) noudattaa autosomaalisesti resessiivisesti periytyvää kaavaa. (Autosomit ovat numeroituja kromosomeja-eli kaikkia kromosomeja lukuun ottamatta X: ää ja Y: tä, jotka määrittävät sukupuolen.)
resessiivisesti periytyvät sairaudet vaativat kaksi geenivirhettä — yleensä yhden kummaltakin vanhemmalta, mutta satunnaisesti yhden toiselta vanhemmalta ja sikiönä esiintyvä tauti on muodostumassa. Ihmiset, joilla on vain yksi geenivirhe resessiiviseen sairauteen, sanotaan kantajiksi, eikä heillä yleensä ole mitään oireita. Usein perheellä ei ole aavistustakaan siitä, että jotkut jäsenet ovat kantajia, ennen kuin lapsi on syntynyt resessiivisen häiriön kanssa.
jos molemmat vanhemmat ovat kromosomissa 5 olevan geenivirheen kantajia, riski siihen, että jokainen raskaus tuottaa lapselle taudin, on 25%. Riski ei muutu, vaikka pariskunnalla olisi kuinka monta lasta.
geneettiset testit kromosomiin 5 liittyvän SMA: n selvittämiseksi on saatavilla niille, joilla epäillään olevan tämä tauti, mukaan lukien syntymättömät vauvat, sekä taudin kantajille. Geenitestaus laajenee ja muuttuu nopeasti, mutta sen vaikutukset voivat olla monimutkaisia. On parasta puhua geneettisen neuvonantajan kanssa ennen testaukseen ryhtymistä. (Geneettinen neuvonta lähete voidaan saada kautta MDA Care Center tai ensisijainen hoito lääkäri).
on välttämätöntä diagnosoida SMA mahdollisimman aikaisin, mieluiten ennen oireiden alkamista, koska hoidon viivästyminen voi pahentaa taudin kulkua ja aiheuttaa pysyviä vaurioita motoneuroneihin. Paras tapa tunnistaa SMA ennen oireiden ilmaantumista on seuloa kaikki vastasyntyneet smn1 exon 7-poiston varalta. Koska Spinraza (nusinersen), tautia muokkaava hoito, hyväksyttiin vuonna 2016 Yhdysvaltain elintarvike-ja lääkevirastossa (FDA) ja varhainen hoito osoitti myönteisiä tuloksia, Yhdysvaltain terveysministeriön sihteeri hyväksyi SMA: n lisäämisen vastasyntyneiden seulontapaneeleihin.1 lisätietoja, käy osoitteessa the hyperlink ” https://strongly.mda.org/sma-added-national-list-disorders-to-screen-for…”SMA lisätty National List of Disorders to Screen for at Birth.
X-linkitetty SMA periytyy X-kromosomin kautta. Naarailla on kaksi X-kromosomia, ja niitä, joilla on geenivirhe yhdessä X-kromosomissa, pidetään yleensä X-linkitetyn taudin kantajina. Uroksilla ei kuitenkaan ole toista X: ää, joka suojaisi niitä geenivirheen kaikilta vaikutuksilta X-kromosomissa ja osoittaisi tällaisen virheen täydet vaikutukset.
lisäksi SMA: ta voivat aiheuttaa mutaatiot dync1h1-geenissä kromosomissa 14. Tämä muoto periytyy dominantisti, eli taudin aiheuttajaksi riittää vain yksi toiselta vanhemmalta periytyvä DYNC1H1-geenimutaatio.
voit lukea lisää SMA: n genetiikasta ja tämän taudin geneettisestä testauksesta, Katso myös:
- MDA Genetic Counseling Webinar vastauksia avainkysymyksiin, Quest News Online, Helmikuu. 28, 2012
- Facts About Genetics and Neuromuscular Diseases, MDA, joulukuu 2009
- The Genie ’ s Out of the Bottle: Genetic testing in the 21st century, Quest Magazine (MDA), marraskuu 2008
- the Pain and Promise of Prenatal and Newborn Genetic Diagnosis, Quest Magazine (MDA), heinäkuu 2007