ATrain Education
Bien que nous en apprenions plus chaque jour sur la physiopathologie de la maladie de Parkinson, elle est toujours considérée comme largement idiopathique (de cause inconnue). Il implique probablement l’interaction de la susceptibilité de l’hôte et des facteurs environnementaux. Un petit pourcentage de cas sont génétiquement liés et les facteurs génétiques font l’objet d’une étude approfondie.
Physiologiquement, les symptômes associés à la maladie de Parkinson sont le résultat de la perte d’un certain nombre de neurotransmetteurs, notamment la dopamine. Les symptômes s’aggravent avec le temps car de plus en plus de cellules touchées par la maladie sont perdues. L’évolution de la maladie est très variable, certains patients présentant très peu de symptômes en vieillissant et d’autres dont les symptômes progressent rapidement.
La maladie de Parkinson est de plus en plus considérée comme une maladie neurodégénérative complexe avec une séquence de progression. Il existe des preuves solides qu’il affecte d’abord le noyau moteur dorsal du nerf vague et les bulbes olfactifs et le noyau, puis le locus coeruleus, et finalement la substantia nigra. Les zones corticales du cerveau sont affectées à un stade ultérieur. Les dommages causés à ces divers systèmes neuronaux expliquent les changements physiopathologiques à multiples facettes qui causent des déficiences non seulement du système moteur, mais aussi des systèmes cognitifs et neuropsychologiques (Kwan & Whitehill, 2011).
Le rôle de la dopamine
La dopamine, comme les autres neurotransmetteurs, transmet des messages chimiques d’une cellule nerveuse à une autre à travers la synapse, un espace entre la cellule présynaptique et le récepteur postsynaptique. La dopamine est sécrétée dans la synapse à partir de vésicules de stockage membranaire dans la membrane présynaptique. Il traverse la synapse et se lie à la membrane postsynaptique, où il active les récepteurs de la dopamine. La dopamine inutilisée restant dans la synapse est de nouveau absorbée dans la cellule présynaptique; une fois de retour dans la cellule présynaptique, l’excès de dopamine est reconditionné dans des vésicules de stockage et libéré une fois de plus dans la synapse.
Dans la synapse, lorsque la dopamine se déplace d’une cellule à l’autre, elle peut être décomposée et rendue inactive par deux enzymes, la MAO (monoamine oxydase) et la COMT (catéchol-O-méthyl transférase). Une stratégie thérapeutique introduit un inhibiteur de la MAO dans la synapse, qui interrompt l’action de l’enzyme MAO et empêche la dégradation de la dopamine. Cela permet à plus de dopamine de rester dans la synapse et augmente la probabilité qu’elle se lie à la membrane postsynaptique.
Transmission synaptique chimique
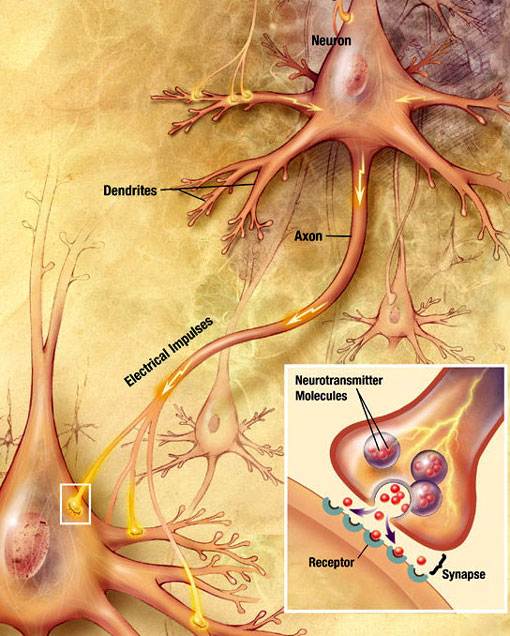
Une onde électrochimique appelée potentiel d’action se déplace le long de l’axone d’un neurone. Lorsque le potentiel d’action atteint le terminal présynaptique, il provoque la libération d’une petite quantité de molécules de neurotransmetteurs, qui se lient à des molécules de récepteurs chimiques situées dans la membrane du neurone postsynaptique, de l’autre côté de la fente synaptique. Source : Wikimedia Commons.
Perte progressive de dopamine
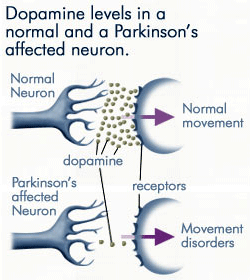
Comme de moins en moins de dopamine est produite par les neurones affectés par la maladie de Parkinson, beaucoup moins de dopamine est disponible pour se lier aux récepteurs de la dopamine sur la membrane post-synaptique. Source : anti-agingfirewalls.com.
Bien que la perte de cellules dopaminergiques ne puisse pas être mesurée directement, des mesures chez des personnes neurologiquement normales et chez des primates non humains révèlent une perte progressive lente de dopamine avec l’âge. Dans la maladie de Parkinson, la perte se produit à un taux beaucoup plus élevé et les mesures biochimiques et les études d’imagerie suggèrent qu’il y a une diminution significative de la dopamine au moment où les symptômes moteurs apparaissent. De ce point de vue, la maladie de Parkinson est une version accélérée de la mort cellulaire observée avec le vieillissement normal (Cookson, 2009). Ceci est illustré dans le graphique ci-dessous, qui montre le déclin des neurones dopaminergiques pendant le vieillissement normal, dans la PD idiopathique, dans la PD causée par des facteurs environnementaux ou génétiques, et dans la PD précoce.
Évolution de la déplétion dopaminergique dans la maladie de Parkinson
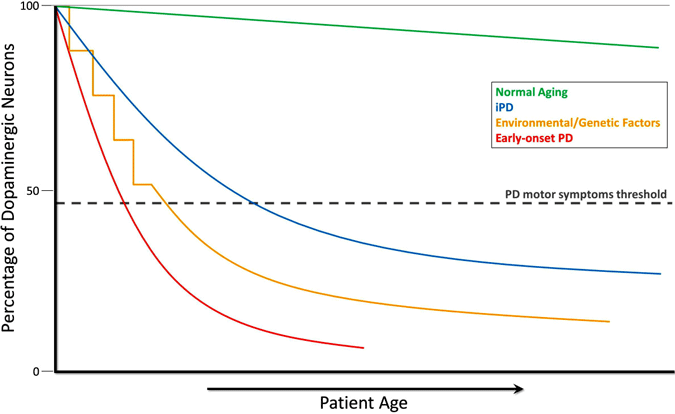
Au cours du vieillissement normal (ligne verte), une dégénérescence dopaminergique petite mais lente se produit sans symptômes moteurs. La PD idiopathique (DPI, ligne bleue) est d’origine inconnue, mais on pense qu’elle se développe progressivement, avec une dégénérescence lente des neurones dopaminergiques conduisant aux symptômes moteurs classiques de la PD plus tard dans la vie. Un autre modèle de neurodégénérescence dopaminergique conduisant à des symptômes moteurs de la MP implique une exposition répétée à des toxiques environnementaux au fil du temps en combinaison avec une prédisposition génétique à la perte de neurones dopaminergiques (ligne jaune). La PD précoce (ligne rouge), causée par des mutations du gène de la PARKINE, implique un déclin précipité des neurones dopaminergiques, et les symptômes moteurs de la PD peuvent survenir des décennies avant ceux de la PD idiopathique. Un autre scénario (non illustré) de développement de symptômes moteurs de la MP implique d’éventuels toxiques environnementaux in utero ou des facteurs génétiques conduisant à un nombre atypiquement faible de neurones dopaminergiques à la naissance et à une susceptibilité accrue au développement de la MP (Haas et al., 2012).
La dégénérescence des neurones dopaminergiques est particulièrement évidente dans une partie de la substance noire appelée pars compacta. De manière significative, la perte de dopamine dans le pars compacta augmente la commande excitatrice globale dans les ganglions de la base, * perturbant le contrôle moteur volontaire et provoquant les symptômes caractéristiques de la MP. La normalisation de la fonction motrice est observée initialement avec le traitement par lévodopa (Gasparini et al., 2013).
* Les principaux composants des ganglions de la base sont le striatum (noyau caudé et putamen), le globus pallidus, la substantia nigra, le noyau accumbens et le noyau sous-thalamique.
À mesure que la gravité de la MP augmente, l’épuisement de la dopamine entraîne d’autres changements dans les voies des ganglions de la base, y compris une modification de la fonction d’autres neurotransmetteurs des ganglions de la base tels que le glutamate, le GABA et la sérotonine (Gasparini et al., 2013). Bien qu’il existe une vulnérabilité relative des neurones producteurs de dopamine dans la substance noire, toutes les cellules dopaminergiques ne sont pas affectées par la maladie de Parkinson; dans certaines parties du cerveau, les neurones producteurs de dopamine sont relativement épargnés (Cookson, 2009).
La voie nigrostriatale
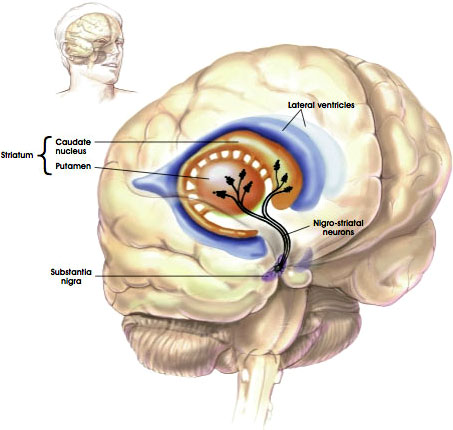
Source: NIH, n.d.
Les corps de Lewy et l’Alpha-synucléine
Les corps de Lewy sont des agrégats anormaux et des inclusions de protéines qui se développent à l’intérieur des cellules nerveuses chez les personnes atteintes de la maladie de Parkinson. Les agrégations sont généralement constituées d’agrégats fibrillaires insolubles contenant des protéines mal repliées. Un grand nombre de molécules ont été identifiées dans les corps de Lewy, mais une protéine appelée alpha-synucléine en est le composant principal.
Corps de Lewy (Inclusions d’alpha-synucléine)
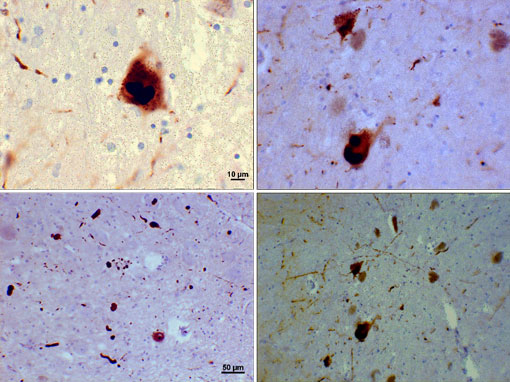
Photomicrographie de régions de substantia nigra chez un patient atteint de Parkinson montrant des corps de Lewy et des neurites de Lewy dans divers grossissements. Les panneaux supérieurs montrent un grossissement de 60x des inclusions intraneuronales de l’alpha-synucléine agrégées pour former des corps de Lewy. Les panneaux inférieurs sont des images à grossissement de 20 × qui montrent des neurites de Lewy ressemblant à des brins et des corps de Lewy arrondis de différentes tailles. Images gracieuseté de Suraj Rajan.
La pathologie de Lewy englobe de nombreuses régions du cerveau et certains rapports ont suggéré que la substantia nigra n’est pas le premier endroit où les corps de Lewy se forment dans la maladie de Parkinson. Les inclusions et les agrégats symbolisent probablement l’étape finale d’une cascade d’événements compliqués. Un stade plus précoce peut être plus directement lié à la pathogenèse du trouble que les inclusions elles-mêmes, qui peuvent ou non représenter des caractéristiques diagnostiques.
Des corps de Lewy sont également observés dans la « démence avec des corps de Lewy », ce qui suggère que ces conditions sont liées les unes aux autres par une pathologie partagée et éventuellement par une étiologie partagée. Ni la perte cellulaire ni la formation de corps de Lewy ne sont absolument spécifiques à la MP, mais les deux sont nécessaires pour un diagnostic de la MP selon les définitions actuelles (Cookson, 2009).
On réalise de plus en plus que les troubles neurodégénératifs tels que la maladie d’Alzheimer, la dégénérescence fronto-temporale, la maladie à prions, la chorée de Huntington et les maladies motoneurones ont des mécanismes cellulaires et moléculaires communs, y compris l’agrégation protéique et la formation de corps d’inclusion dans certaines zones du système nerveux (Jellinger, 2011).
Inflammation et réponse immunitaire
Le déclencheur de la dégénérescence dopaminergique semble être multifactoriel – affecté par des éléments endogènes et environnementaux. L’inflammation et les réponses immunitaires sont de plus en plus considérées comme des médiateurs importants de la dégénérescence dopaminergique. Des études de grande population ont suggéré que les personnes prenant des anti-inflammatoires non stéroïdiens (AINS) ont moins de risque de développer une MP idiopathique, ce qui suggère que les anti-inflammatoires peuvent être un traitement modificateur de la maladie prometteur pour les patients parkinsoniens (Barcia, 2013).
De nouvelles phases d’essai ont impliqué des traitements anti-inflammatoires – en particulier la recherche d’un biomarqueur objectif dans les traitements visant à réduire les modifications inflammatoires chez les patients atteints de MP. Les chercheurs utilisent des outils de neuroimagerie pour développer un biomarqueur pertinent dans le but de le tester dans de grands essais d’imagerie clinique. Les résultats de ces essais fourniront des données pour tester et surveiller la progression des traitements anti-inflammatoires de la MP et aideront à identifier la fenêtre thérapeutique opportune pour arrêter, ou du moins ralentir, la dégénérescence dopaminergique à médiation inflammatoire (Barcia, 2013).
Parkinsonisme
Le parkinsonisme, également appelé « Parkinson atypique », « Parkinson secondaire » ou « syndrome de Parkinson », est un syndrome neurologique dans lequel un patient présente certains des symptômes associés à la maladie de Parkinson — tremblement, rigidité, bradykinésie et instabilité posturale. Mais le parkinsonisme n’est pas la maladie de Parkinson. On ne pense pas que le parkinsonisme soit causé par la maladie de Parkinson et les patients répondent généralement mal à l’intervention pharmacologique. Le parkinsonisme a souvent une cause identifiable, telle qu’une exposition à des toxines, à la méthamphétamine, à un traumatisme, à plusieurs accidents vasculaires cérébraux, à d’autres troubles du système nerveux ou à une maladie. Généralement, les corps de Lewy ne sont pas vus dans le parkinsonisme.
Le terme parkinsonisme est également associé à des troubles tels que la paralysie supranucléaire progressive, l’atrophie systémique multiple, la démence à corps de Lewy, la dégénérescence corticobasale, le parkinsonisme vasculaire, le parkinsonisme induit par un médicament et le parkinsonisme secondaire à une infection et à d’autres causes (Hohler et al., 2012). Une forme de parkinsonisme réversible peut survenir lors de l’utilisation de certains médicaments neuroleptiques, en particulier la réserpine, les antipsychotiques (halopéridol) et le métoclopramide. L’exposition à certaines toxines, une intoxication grave au monoxyde de carbone et une intoxication au mercure peuvent également entraîner un parkinsonisme.
L’apparition au début des années 1980 de symptômes de parkinsonisme chez un groupe de toxicomanes ayant consommé un lot contaminé d’un opiacé synthétique a conduit à la découverte du produit chimique MPTP comme agent responsable du syndrome de parkinsonisme chez les primates non humains ainsi que chez les humains. Le MPTP peut être produit lors de la fabrication d’une forme d’héroïne (le MPTP est converti en une neurotoxine qui détruit sélectivement les cellules dopaminergiques de la substance noire). Ces cas sont rares et ont surtout affecté les consommateurs de drogues à long terme.
L’abus de méthamphétamine a également été lié au parkinsonisme. Chez les animaux de laboratoire, l’exposition à la méthamphétamine endommage les fibres dopaminergiques du striatum* ainsi que les corps cellulaires de la substance noire, faisant écho à la dégénérescence observée chez les patients humains atteints de MP. Des dommages sélectifs aux terminaux dopaminergiques du striatum ont également été observés chez des utilisateurs humains de méthamphétamine, bien qu’il n’y ait aucune preuve jusqu’à présent que l’abus de méthamphétamine endommage les corps cellulaires dopaminergiques de la substance noire (Granado et al., 2013).
* Le plus gros noyau des ganglions de la base, le striatum est constitué du noyau caudé et du putamen.
On a émis l’hypothèse que la consommation de méthamphétamine pourrait prédisposer les utilisateurs au développement futur de la MP. Cette hypothèse a été étayée par des travaux épidémiologiques récents indiquant que les utilisateurs de méthamphétamine ont un risque accru de développer une MP. Cela concorde avec les effets neurotoxiques persistants de la méthamphétamine chez les animaux de laboratoire (Granado et al., 2013).
Les patients atteints de parkinsonisme sont souvent difficiles à gérer en ambulatoire. La complexité de leurs symptômes, les déficits cognitifs et autonomes supplémentaires, la mauvaise réponse à la plupart des médicaments contre la MP et le déclin relativement rapide de l’état contribuent aux défis de la prise en charge de ces patients, en particulier à mesure que la maladie progresse (Hohler et al., 2012).