Chimie bioorganique et organique
Réaction d’hydroboration-oxydation
L’addition cis de diborane à une liaison alcène fournit une méthode d’hydratation extrêmement utile. Le diborane peut être généré par addition de borohydrure de sodium à de l’éthérate de trifluorure de bore dans du tétrahydrofurane ou de l’éther à 0o-5oC. Le diborane est le dimère du borane (BH3) et est une forme stable de ce réactif (Scheme1).
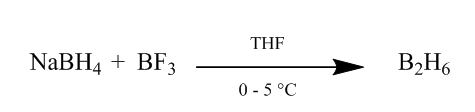
Figure 1 Obtention de Diborane à partir de borohydrure de sodium
L’addition de diborane à l’alcène est extrêmement rapide et généralement, le réactif s’ajoute à partir de la moins encombrée des deux faces du système π. L’addition cis a été rationalisée par un état de transition à quatre centres. Le complexe borane résultant de l’addition de diborane à un alcène est converti, avec rétention de stéréochimie, en un alcool par traitement au peroxyde d’hydrogène basique. Ainsi le 1-méthylcyclohexène 1 lors de l’hydroboration-oxydation conduit à la formation du trans-2-méthylcyclo-hexanol 2. Le chemin mécaniste a été décrit dans (Schéma 2). La méthode de conversion d’alcène en alcool par hydroboration-oxydation a été appliquée pour la synthèse de nombreux produits naturels. Quelques exemples sont illustrés ci-dessous.

Figure 2 Mécanisme de réaction pour la formation du trans-2-méthylcyclo-hexanol 2
Synthèse de (±) junénol et de (±) acalomone
L’utilisation de la réaction d’hydroboration-oxydation a été observée par Banerjee et ses collègues lors de la synthétise3 des sesquiterpènes d’eudesmone (±) junénol et (±) acalomone. Pour réaliser la synthèse de ces sesquiterpènes, l’alcène 3, a été choisi comme matière première et soumis à une hydroboration-oxydation pour conduire à l’alcool 4 (Schéma 3). La cétone 5, obtenue par oxydation de l’alcool avec le réactif de Jones 4, a été mise en réaction avec le carbonate de diéthyle. Le produit obtenu est traité avec du méthyl-lithium pour obtenir le cétol 6 dont la conversion en isopropyl-cétone 7 s’effectue respectivement par déshydratation et hydrogénation. Une réduction de la cétone par hydrure métallique suivie d’une oxydation par le tétraacétate de plomb 5 dans le cyclohexane conduit à l’éther cyclique 8, qui est transformé en acide cétoacide 9 par oxydation par l’acide chromique et l’acide acétique. La décarboxylation avec le tétraacétate de plomb dans le benzène et la pyridine suivie d’une purification sur gel de silice imprégné d’AgNO3 à 10% donne (±) acolamone 10. Une réduction de l’acolamone 10 par le borohydrure de sodium dans le méthanol, suivie d’une sublimation du produit obtenu, conduit au junénol 11.
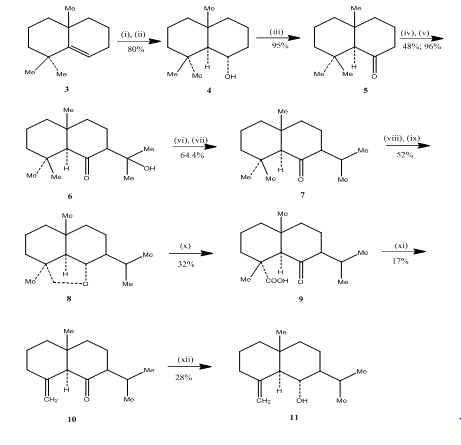
Figure 3 Synthèse des réactifs eudesmonés sesquiterpènes (±) junénol et (±)-acalomone
: (i) BF3.Et2O, NaBH4, THF, 0-5 °C; (ii) NaOH (10%), H2O2 (30%); (iii) CrO3/HMPT; (iv) NaH, CO (OEt) 2, DME; (v) MeLi, Et2O, reflux, 2h; (vi) HCl (conc), MeOH; (vii) H2, PtO2, MeOH; (viii) Na, EtOH, reflux; (ix) Pb (CAo) 4, C6H12; (x) CrO3, AcOH; (xi) Pb (CAo) 4, C6H6, Py, reflux; (xii) NaBH4, EtOH.
Synthèse de l’acide pisiférique
L’utilisation de l’hydroboration-oxydation a été enregistrée lors de la synthèse de l’acide pisiférique,6 un diterpène tricyclique qui présente des activités antibactériennes contre toutes les bactéries à gram positif testées.7 La voie synthétique a été représentée dans le schéma 4. L’hydroboration-oxydation de l’alcène 13, préparé à partir du cétoalcool 12 connu 8, a été oxydée avec le réactif de jones 4 et réduite respectivement avec de l’hydrure métallique pour donner l’alcool 14. L’oxydation avec du tétraacétate de plomb dans le benzène avec une lampe au tungstène de 250W a donné l’éther cyclique 15. Le clivage de l’éther cyclique avec le zinc, l’iodure de zinc et l’acide acétique8 fournit le pisiférol 16. La transformation du pisiférol en ester 17 a été réalisée en six étapes:
- Méthylation au sulfate de diméthyle
- Oxydation au réactif de jones
- Estérification au diazométhane
- Réduction au borohydrure de sodium
- Tosylation
- Détosylation
L’ester l7 a été transformé en acide pisiférique 18 par chauffage avec du bromure d’aluminium et de l’éthane thiol.
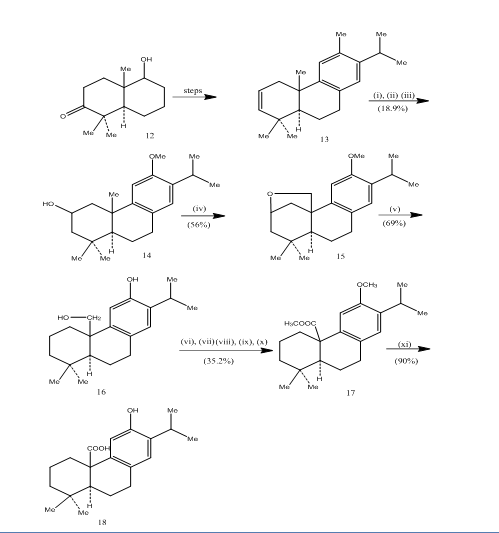
Figure 4 Synthèse des réactifs de l’acide pisiférique 18
: (i) BF3.Et2O, NaBH4; (ii) NaOH (10%), H2O2 (30%), H2SO4-HCrO4; (iii) LiAlH4, THF; (iv) Lampe au tungstène Pb (OAc) 4, CaCO3, C6H6, 250w; (v) Zn, ZnI, MeCOOH; (vi) MéSO4, Me2CO; (vii) H2SO4-HCrO4; (viii) CH2N2, Et2O; (ix) NaBH4, MeOH; (x) TsCl, Py; (xi) NaI, poussière de Zn, DMF; (xii) AlBr3, (CH2SH) 2.
La réaction d’hydroboration-oxydation a été appliquée pour la synthèse de (±)eudes-4(14),7(11)- diène-8-one, 9 taxodiones, 10 alcools de norditerpène11 et de nombreux autres terpènes.12 Ces exemples indiquent clairement l’utilisation de l’éthérate de tifluorure de bore dans la conversion des alcènes en alcools et par la suite leurs transformations en composés terpénoïdes.
Clivage des époxydes
Les époxydes peuvent être clivés par plusieurs réactifs. L’éthérate de borontrifluorure d’acide de Lewis a également été utilisé pour le clivage des époxydes et, dans de nombreux cas, le produit résultant se réarrange en cétone. Le clivage des époxydes s’accompagne également d’une cyclisation. Dans cette revue, le clivage de certains époxydes avec l’éthérate de trifluorure de bore et l’utilisation des produits résultants dans la synthèse de produits naturels ont été discutés.
Synthèse de la 6-méthoxy-2-tétralone
Le clivage de l’époxyde avec l’éthérate de trifluorure de bore a été utilisé13 pour la synthèse de la 6-méthoxy-2-tétralone 20 (Schéma 5), un matériau de départ important pour la synthèse de nombreux composés organiques. L’époxydation de l’alcène13 19 suivie d’un traitement du produit brut dans le dichlorométhane par de l’éthérate de trifluorure de bore a permis d’obtenir la tétralone 20 avec un rendement de 36%. Lorsque le clivage a été essayé avec de l’acide sulfurique, le rendement de la teralone 20 a été amélioré (39%) ainsi que la formation d’autres produits secondaires et la purification chromatographique a donc été très laborieuse.
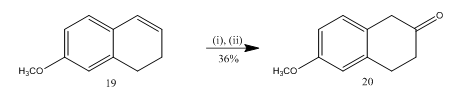
Figure 5 Synthèse des réactifs 6-méthoxy-2-tétralone 20
: (i) MCPBA, CH2Cl2; (ii) BF3OEt2
Synthèse du cuprane
Le réarrangement des époxydes par l’éthérate de trifluorure de bore s’est avéré très utile lors de la synthétise14 du cuprane de sesquiterpène. La voie synthétique est décrite dans le schéma 6. Le 6,6-diméthyl-1-p-tolylcyclohexène 21 lors de l’époxydation a donné un bon rendement à l’époxyde 22 qui, après traitement par de l’éthérate de trifluorure de bore dans le benzène, a donné l’aldéhyde 23 avec un faible rendement. La semicarbazone de l’aldéhyde a été chauffée avec de l’hydroxyde de potassium pour fournir le cuprane sesquiterpénique 24 avec un rendement acceptable. La synthèse est attrayante en raison de sa brièveté dans les étapes. Les conditions utilisées pour le réarrangement de l’époxyde 22 sont critiques car il a tendance à subir un réarrangement supplémentaire en cétone 25.
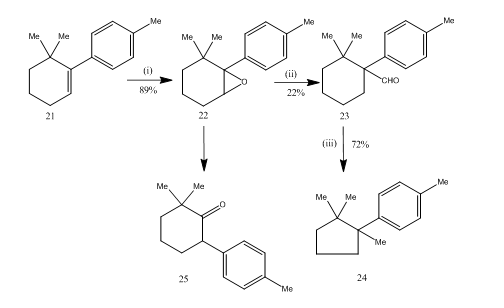
Figure 6 Synthèse des réactifs cuprane
: (i) PhCO3H, CHCl3 ; (ii) C6H6, BF3Et2O ; (iii) NH2NHCONH2, KOH
La synthèse de (±)éther de karahana
Éthérate de trifluorure de bore a également été utilisée pour le clivage de l’époxyde lors de la synthétise15 de l’éther de karahana, un monoterpène volatil qui a été isolé16 du houblon japonais. La voie synthétique est décrite dans le schéma 7. L’époxyde 27, obtenu à partir du diène 26, après traitement par de l’éthérate de trifluorure de bore a subi une cyclisation conduisant au produit 28. La cyclisation s’est probablement produite par l’intermédiaire 27 (i). La réduction de l’hydrure métallique donne du diol qui, lors de la tosylation, donne du karahanaéther 29. Le rendement n’est pas spécifié. Le clivage des époxydes a été utilisé pour la synthèse de nombreux terpènes comme la rosénolactone, la cypérolone 17, le maritimol 18.19
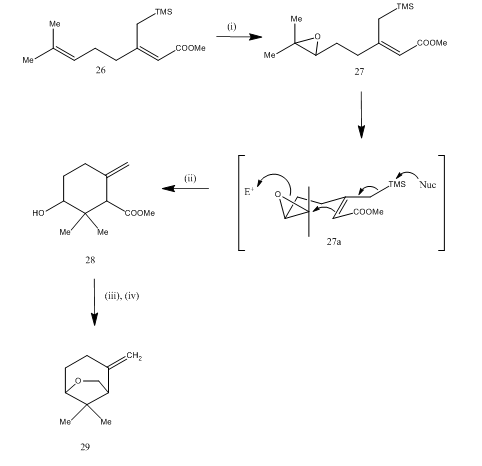
Figure 7 Synthèse de réactifs (±)éther de Karahana
: (i) MCPBA; (ii) BF3Et2O; (iii) LiAlH4; (iv) TsCl, Py
Estérification
L’estérification est une réaction fréquemment utilisée pour la synthèse de nombreux composés organiques. L’éthérate–alcool de trifluorure de bore est un réactif très pratique pour l’estérification de nombreux acides p-amino benzoïques, acides aromatiques, hétérocycliques et insaturés.20 Dans certaines réactions d’estérification, l’utilisation de ce réactif a fourni un rendement supérieur par rapport à d’autres réactifs. Quelques exemples sont donnés dans le schéma 8. Les acides 30-32 ont été convertis en esters 33-35 respectivement avec un rendement élevé sur traitement avec un réactif éthérate-alcool de trifluorure de bore. Marshall et ses collaborateurs21 ont utilisé le même réactif pour l’estérification des acides carboxyliques. Dymicky22 a préparé plusieurs formats à haut rendement à partir d’acide formique et d’alcool en présence d’une quantité catalytique de complexe trifluorure de bore-méthanol. Les autres catalyseurs, par exemple l’acide sulfurique, l’acide p-toluène sulfonique, n’étaient pas aussi efficaces que le complexe trifluorure de bore-méthanol.
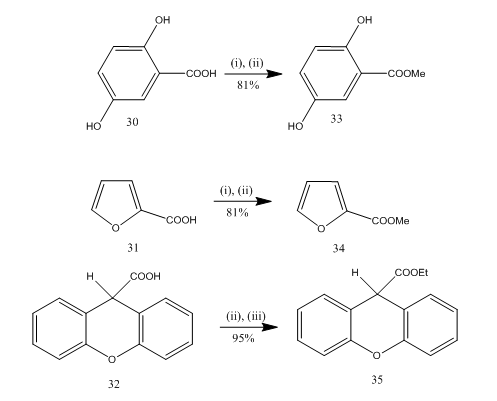
Figure 8 Estérification des acides 32-35
Réactifs : (i) MeOH; (ii) BF3.Et2O; iii) EtOH
Jackson et ses collaborateurs 23 ont mis au point une méthode efficace de conversion des alcools 37-39 et des acides 40-42 directement en dérivés t-butyliques correspondents avec un bon rendement en utilisant le trichloroacétimidate de t-butyle 36 en présence d’une quantité catalytique d’éthérate de trifluorure de bore telle que présentée dans le schéma 9. Cette méthode fonctionne mieux avec les groupes sensibles à l’acide que les méthodes traditionnelles utilisant l’isobutène. Un groupe hydroxyle moins encombré d’un diol peut être protégé et se prête également à un travail à petite échelle (en évitant la manipulation d’isobutène gazeux). Le 2,2,2-trichloroacétimidate de t-butyle 36 est facilement préparé par addition de t-butanol sur le trichloroacétonitrile. La plupart des expériences ont été réalisées en présence d’un mélange de dichlorométhane et de cyclohexane. L’anhydride acétique en présence d’éthérate de trifluorure de bore a été utilisé pour l’acétylation du groupe hydroxyle.24

Figure 9 Conversion d’alcools et d’acides à partir de dérivés du t-butyle.
Réactifs : 36, (i) BF3.Et2O, (ii) CH2Cl2, C6H12
Cyclisation
L’éthérate de trifluorure de bore a joué un rôle important dans la cyclisation de nombreux acides carboxyliques, allènes, etc. Les quelques exemples suivants illustreront le rôle de l’éthérate de trifluorure de bore comme agent cyclisant. Le chlorure d’acide 49 et l’alcène 50 ont été condensés pour conduire à la cétone divinyl25 51 qui a subi une cyclisation de nazarov26,27 fournissant la cétone cyclique 52 qui a été convertie en trichodiène sesquiterpène 53 (Schéma 10).
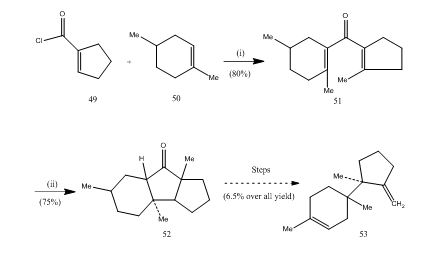
Figure 10 Synthèse des réactifs du sesquiterpène trichodiène 53
: (i) SnCl4, NaOMe; (ii) BF3Et2O
Plusieurs cétones d’allénylaryl subissent une cyclisation avec de l’éthérate de trifluorure de bore donnant de la méthylène benzocyclopenténone via une nouvelle cyclisation en mode 5-endo.28 Les cétones 54-56 ont donné respectivement des benzocyclopenténones 57-59 (Schéma 11). La transformation s’est probablement produite comme le montre la cyclisation de l’allénylarylcétone 54 en 57. On peut observer que la présence de groupements substituants dans le cycle aromatique détermine le rendement du produit cyclisé. Kos et Loewenthal28 ont rapporté la cyclisation de l’acide 60 avec de l’éthérate de trifluorure de bore en cétone 61 qui a été transformée en gibbérone 62 (Schéma 12) en trois étapes:
- Cétalisation
- Réduction de Huang-Minlon et
- Hydrolyse acide. Les exemples mentionnés ci-dessus montrent l’utilisation de l’éthérate de trifluorure de bore dans la cyclisation de composés organiques
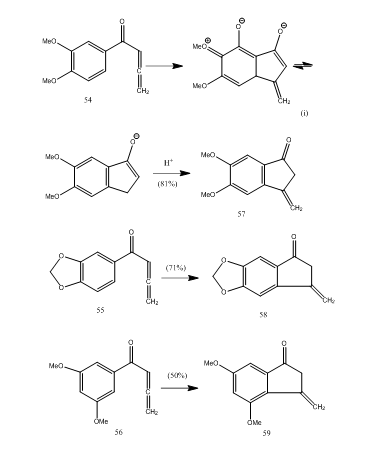
Figure 11 Synthèse des Cyclopenténones

Figure 12 Synthèse en réactifs de la gibbérone
: (i) BF3.Et2O; (ii) (a) C2H6O2; (b) DEG, N2H4, KOH, 190-200 ° C; (c) H+