Paralysies périodiques
Une classification cliniquement utile des paralysies périodiques primaires, présentée dans le tableau 1, comprend les formes hypokaliémiques, hyperkaliémiques et paramyotoniques.
Tableau 1. Paralysie périodique primaire (modifiée à partir de Jurkat-Rott et Lehmann-Horn) (Ouvrir le tableau dans une nouvelle fenêtre)
|
Maladie |
Gene |
Protéines |
Héritage |
Mutation |
|
HyperPP |
SCN4A |
Nav1.4 |
Dominante |
Bien |
|
NormoPP |
Fine (ω-pore) |
|||
|
Paramyotoniecongénita |
Bien |
|||
|
HypoPP Type II |
Fine (ω-pore) |
|||
|
HypoPP Commande À |
CACNA1S |
Cav1.1 |
Dominante |
Gain (ω-pore) |
|
ThyrotoxicPP |
KCNJ18 |
Kir2.18 |
Dominante |
Perte |
|
Syndrome d’Andersen-Tawil |
KCNJ2 |
Kir2.1 |
Dominante |
Perte |
La base physiologique de la faiblesse flasque est l’inexcitabilité de la membrane musculaire (c’est-à-dire le sarcolemme). L’altération du taux de potassium sérique n’est pas le principal défaut de la PP primaire; l’altération du métabolisme du potassium est le résultat de la PP. Dans la PP primaire et thyrotoxique, une paralysie flasque se produit avec des changements relativement faibles du taux de potassium sérique, tandis que dans la PP secondaire, les taux de potassium sérique sont nettement anormaux.
Aucun mécanisme unique n’est responsable de ce groupe de troubles. Ainsi, ils sont hétérogènes mais partagent quelques traits communs. La faiblesse est généralement généralisée mais peut être localisée. La musculature crânienne et les muscles respiratoires sont généralement épargnés. Les réflexes d’étirement sont absents ou diminués pendant les attaques. Les fibres musculaires sont électriquement inexcitables lors des attaques. La force musculaire est normale entre les attaques mais, après quelques années, un certain degré de faiblesse fixe se développe dans certains types de PP (en particulier les PP primaires). Toutes les formes de PP primaires (à l’exception de la myotonie congénitale de Becker) sont héréditaires autosomiques dominantes ou sporadiques (résultant probablement de mutations ponctuelles).
Les canaux ioniques sensibles à la tension régulent étroitement la génération de potentiels d’action (altérations brèves et réversibles de la tension des membranes cellulaires). Ce sont des canaux ioniques sélectivement et variablement perméables. Les transporteurs d’ions dépendants de l’énergie maintiennent des gradients de concentration. Pendant la génération des potentiels d’action, les ions sodium se déplacent à travers la membrane à travers des canaux ioniques à chaîne de tension. La membrane des fibres musculaires au repos est polarisée principalement par le mouvement du chlorure à travers les canaux de chlorure et est repolarisée par le mouvement du potassium. Les channelopathies de sodium, de chlorure et de calcium, en tant que groupe, sont associées à la myotonie et à la PP. Les sous-unités fonctionnelles des canaux sodiques, calciques et potassiques sont homologues. Les channelopathies de sodium sont mieux comprises que les channelopathies de calcium ou de chlorure. Toutes les formes de PP familiales montrent la voie mécaniste finale impliquant une dépolarisation aberrante, des canaux sodiques inactivants et une inexcitabilité des fibres musculaires.
La discussion dans cet article porte principalement sur les channelopathies sodiques, calciques et potassiques ainsi que sur les formes secondaires de PP. Les channelopathies de chlorure ne sont pas associées à une faiblesse épisodique et sont discutées plus en détail dans les articles sur les troubles myotoniques.
Résumé du dysfonctionnement du canal dans divers types de PP
Avec l’inactivation rapide du canal HyperPP, les mutations sont généralement situées dans les parties internes des segments transmembranaires ou dans les boucles intracellulaires affectant les sites d’amarrage de la particule inactivante rapide, altérant ainsi l’inactivation rapide du canal conduisant à un courant Na + persistant.
Avec une fuite de cations activée par hyperpolarisation en HypoPP neutralisant le courant de rectification K+, les mutations provoquent une substitution extrême de l’arginine ou de la lysine.
Avec une fuite de cations activée par la dépolarisation NormoPP, les mutations se trouvent dans des emplacements plus profonds du capteur de tension du domaine II au codon R675.
Le dysfonctionnement des canaux ioniques est généralement bien compensé par une excitation normale, et des déclencheurs supplémentaires sont souvent nécessaires pour produire une inexcitabilité musculaire en raison d’une dépolarisation membranaire prolongée.
L’apport en glucose et en potassium a les effets opposés dans ces troubles. En HyperPP, l’apport en potassium déclenche l’attaque, tandis que le glucose l’améliore. En revanche, le glucose provoque des crises hypokaliémiques et le potassium est le traitement de l’attaque.
Notez l’image ci-dessous.
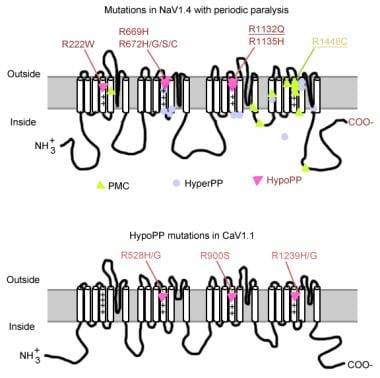 Mutations dans la paralysie périodique.
Mutations dans la paralysie périodique. Gène du canal sodique musculaire
Le canal sodique a une sous-unité alpha et une sous-unité bêta. La sous-unité alpha du canal sodique est une glycoprotéine de 260 kd comprenant environ 1800-2000 acides aminés. Ce canal est très conservé au cours de l’évolution de la Drosophile à l’homme. Il a 4 domaines homologues (I-IV) qui se replient pour former un pore central, chacun avec 225-325 acides aminés. Chaque domaine est constitué de 6 segments hydrophobes (S1-S6) traversant la membrane cellulaire. Les principales fonctions du canal comprennent la gâchette sensible à la tension, l’inactivation et la sélectivité des ions. La boucle extracellulaire entre S5 et S6 plonge dans la membrane plasmique et participe à la formation du pore. Le segment S4 contient des acides aminés chargés positivement à chaque troisième position et fonctionne comme un capteur de tension. Des changements de conformation peuvent survenir pendant la dépolarisation, entraînant l’activation et l’inactivation du canal. La boucle cellulaire entre le domaine III-S6 et le domaine IV-S1 agit comme une porte inactivante.
Le canal sodique a 2 portes (activation et inactivation) et peut exister dans 3 états. Au repos avec la membrane polarisée, la porte d’activation est fermée et la porte d’inactivation est ouverte. Avec la dépolarisation, la porte d’activation s’ouvre, permettant aux ions sodium de traverser le canal ionique et exposant également un site d’amarrage pour la porte d’inactivation. Avec une dépolarisation continue, la porte d’inactivation se ferme, bloquant l’entrée du sodium dans la cellule et provoquant l’entrée du canal dans l’état d’inactivation rapide. Cette inactivation du canal permet à la membrane de se repolariser, entraînant un retour à l’état de repos avec la porte d’activation fermée et la porte d’inactivation ouverte. Deux processus d’inactivation se produisent dans le muscle squelettique des mammifères: l’inactivation rapide implique la fin du potentiel d’action et agit sur une échelle de temps de la milliseconde. L’inactivation lente prend quelques secondes à quelques minutes et peut réguler la population de canaux sodiques excitables.
Les mutations des canaux sodiques qui perturbent l’inactivation rapide et lente sont généralement associées à un phénotype d’HyperPP et de myotonie, où les mutations qui améliorent l’inactivation lente ou rapide produisant une perte de la fonction des canaux sodiques provoquent une HypoPP.
Les mutations du gène du canal sodique (SCN4A) présentent plusieurs caractéristiques générales. La plupart des mutations se trouvent dans le lieur « inactivant » entre les répétitions III et IV, dans le segment « sensible à la tension » S4 de la répétition IV ou au niveau de la membrane interne où elles pourraient altérer le site d’amarrage de la grille d’inactivation. Le phénotype clinique diffère par une substitution spécifique d’acides aminés et, bien qu’un certain chevauchement puisse se produire entre la PP hyperkaliémique, la paramyotonie congénitale (PC) et les myotonies aggravées par le potassium (PAM), les 3 phénotypes sont généralement distincts (comme décrit ci-dessous). Presque tous les canaux mutants ont altéré l’inactivation rapide du courant sodique. La plupart des patients sont sensibles au potassium systémique ou à la température froide.
Il existe deux populations de canaux, mutants et sauvages; l’inactivation rapide altérée entraîne une dépolarisation prolongée des membranes des fibres musculaires mutantes et peut expliquer les 2 symptômes cardinaux de ces troubles, la myotonie et la faiblesse. Dans le PP hyperkaliémique, un gain de fonction se produit dans le déclenchement des canaux mutants, entraînant une augmentation du courant de sodium dépolarisant excessivement le muscle affecté. Une légère dépolarisation (5-10 mV) de la membrane des myofibres, qui peut être causée par une augmentation des concentrations extracellulaires de potassium, entraîne le maintien des canaux mutants en mode non inactivé. Le courant de sodium persistant vers l’intérieur provoque une mise à feu répétitive des canaux sodiques de type sauvage, ce qui est perçu comme une raideur (c’est-à-dire une myotonie).
Si une dépolarisation plus sévère (20-30 mV) est présente, les canaux normaux et anormaux sont fixés dans un état d’inactivation, provoquant une faiblesse ou une paralysie. Ainsi, des différences subtiles de gravité de la dépolarisation membranaire peuvent faire la différence entre la myotonie et la paralysie. La sensibilité à la température est une caractéristique du PC. Le froid exacerbe la myotonie et induit une faiblesse. Un certain nombre de mutations sont associées à cette condition, dont 3 au même site (1448) dans le segment S4. Ces mutations remplacent l’arginine par d’autres acides aminés et neutralisent cette charge positive S4 hautement conservée. Les mutations de ces résidus sont la cause la plus fréquente de PC. Certains des mécanismes possibles responsables de la sensibilité à la température sont les suivants:
-
La température peut affecter de manière différentielle le changement conformationnel dans le canal mutant.
-
Des températures plus basses peuvent stabiliser les canaux mutants dans un état anormal.
-
Les mutations peuvent modifier la sensibilité du canal à d’autres processus cellulaires, tels que la phosphorylation ou les seconds messagers.
La plupart des cas de PP hyperkaliémiques sont dus à 2 mutations dans SCN4A, T704M et M1592V. Des mutations dans le canal sodique, en particulier au niveau des résidus 1448 et 1313, sont responsables de la paramyotonie congénitale. Une faible proportion des cas de paralysie périodique hypokaliémique sont associés à des mutations au niveau des codons 669 et 672 (HypoPP2). En HypoPP2, les mutations des canaux sodiques améliorent l’inactivation pour produire une perte nette de défaut fonctionnel.
Le PP normokaliémique ressemble à la fois à l’HyperPP (sensibilité au potassium) et à l’HypoPP (durée des attaques) et est causé par des mutations SCN4A à un emplacement plus profond du capteur de tension DII au codon 675. Les mutations R675 diffèrent de l’HypoPP en ce que ces mutations entraînent un pore de déclenchement activé par la dépolarisation générant un courant ω avec une dépendance à la tension inversée, car ce site est exposé à des sites extracellulaires lors d’une dépolarisation plus forte.
Gène du canal calcique
Le gène du canal calcique (CACNL1A3) est un complexe de 5 sous-unités (alpha-1, alpha-2, bêta, gamma et delta). Le récepteur de la dihydropyridine du muscle squelettique (DHP) est situé principalement dans la membrane tubulaire transversale. La sous-unité alpha-1 a des sites de liaison pour les médicaments DHP et conduit le courant de calcium lent de type L. Il participe également au couplage excitation-contraction (EC) et agit comme un capteur de tension grâce à sa liaison avec le récepteur de la ryanodine du réticulum sarcoplasmique (c’est-à-dire un canal de libération de calcium). Toute modification du potentiel membranaire est liée à la libération intracellulaire de calcium, ce qui permet le couplage EC. Des mutations ponctuelles dans la sous-unité alpha-1 du récepteur DHP / canal calcique provoquent une hypokaliémie PP (HypoPP1). Deux mutations du gène CACNA1S, R528H et R1239H, sont responsables de la plupart des cas de PP hypokaliémiques.
La base physiologique de la maladie n’est toujours pas comprise, mais est plus probablement due à un échec de l’excitation plutôt qu’à un échec du couplage EC. Cependant, la dépolarisation induite par l’hypokaliémie peut réduire la libération de calcium, affectant directement ou indirectement le contrôle de la tension du canal par inactivation du canal sodique. L’insuline et l’adrénaline peuvent agir de la même manière. Les mutations du gène du canal calcique présentent certaines similitudes avec les mutations SCN4A. Les mutations modifient l’inactivation du canal mais pas l’activation dépendante de la tension. Les enregistrements de cultures de myotube de patients atteints ont révélé une réduction de 30% du courant calcique de type L sensible à la DHP. Les canaux sont inactivés à de faibles potentiels membranaires.
Les mutations des canaux calciques provoquent une perte de fonction qui se manifeste par une densité de courant réduite et une inactivation plus lente. La façon dont cette inactivation est liée aux crises induites par l’hypokaliémie n’est pas comprise. Au moins dans la mutation R528H, une canalopathie secondaire possible se produit, liée à une réduction du courant potassique sensible à l’ATP due à une homéostasie calcique altérée. Les courants plus faibles associés aux mutations CACNL1A3 pourraient légèrement modifier l’homéostasie calcique intracellulaire, ce qui pourrait affecter les propriétés et l’expression des canaux K+, en particulier KATP (canal potassique sensible à l’ATP) appartenant à la classe des canaux redresseurs vers l’intérieur. L’insuline agit également en HypoPP en réduisant ce courant K + redresseur vers l’intérieur.
La perte de charge du capteur de tension représente la plupart des cas d’HypoPP. Les canaux sodiques et calciques ont des sous-unités alfa homologues formant des pores. Les mutations ponctuelles de CACNL1A3 et SCN4A affectent les résidus argentins dans les capteurs de tension S4 de ces canaux. Les mutations de l’arginine dans les segments S4 sont responsables de 90% des cas d’HypoPP.
La perte de charge du capteur de tension représente la plupart des cas d’HypoPP. Les canaux sodiques et calciques ont des sous-unités α homologues formant des pores. Presque toutes les mutations de Cav1.1 (HypoPP-1) et Nav1.4 (HypoPP-2) neutralisent un acide aminé chargé positivement dans l’une des arginines ou lysines les plus externes des capteurs de tension. Le Nav1.4 mutations sont le plus souvent situées dans les capteurs de tension des répétitions I, II et III, provoquant une fuite de cations.
La substitution de l’arginine la plus externe par un acide aminé plus petit tel que la glycine ouvre une voie conductrice à un potentiel hyperpolarisé, ce qui entraîne un courant de cations vers l’intérieur (fuite de cations ou courant ω à distinguer de (ω-) à travers un pore conducteur d’ions, est un courant activé par l’hyperpolarisation de cations monovalents à travers un pore de déclenchement S4 contrecarrant les courants K + rectifiants) dépolarisant ou déstabilisant le potentiel de repos.
Le segment S4 se déplace vers l’extérieur pendant la dépolarisation fermant la voie conductrice. Les fibres musculaires présentant des mutations sévères du capteur de tension sont dépolarisées non seulement pendant l’hypokaliémie, mais également à des niveaux de potassium dans la plage normale, ce qui explique une faiblesse interictale et permanente. Une myopathie sévère avec remplacement graisseux du tissu musculaire est couramment observée chez les patients atteints de Cav1.1 R1239H (mutations DIV).
Les glucocorticostéroïdes provoquent une HypoPP en stimulant la Na + K + ATPase médiée par l’insuline et l’amyline.
Gène des canaux potassiques
La rectification vers l’intérieur est une propriété importante des canaux Kir. La rectification implique un blocage des pores par conduction dépendant de la tension avec des polyamines et du Mg ++ pendant la dépolarisation, et ce blocage est éliminé lors du gradient de potentiel lors de l’hyperpolarisation. Des mutations des canaux potassiques sont observées dans le syndrome d’Andersen-Tawil et la PP thyrotoxique.
La triade de caractéristiques dysmorphiques, de paralysie périodique et d’arythmies cardiaques caractérise le syndrome d’Andersen-Tawil. Ce syndrome est associé à des mutations du gène KCNJ2. Le gène KCNJ2 code pour le canal potassique redresseur vers l’intérieur Kir2.1. Des mutations des canaux potassiques dans le KCNE3 sont rapportées pour provoquer une PP hypokaliémie, mais cela n’a pas été corroboré.
Les mutations de Kir2.6 provoquent une sensibilité au PP thyréotoxique. La faiblesse épisodique observée dans la PP thyrotoxique est similaire à celle observée dans l’HypoPP et le syndrome d’Andersen-Tawil. Ce trouble est le plus répandu chez les Asiatiques et les hommes latino-américains. La PP thyrotoxique est une maladie génétique démasquée par la thyrotoxicose. Kir2.6 est principalement exprimé dans le muscle squelettique. La triiodothyronine améliore la transcription de KCNJ18, ce qui peut entraîner une expression accrue de Kir2.6. La PKC est activée pendant la thyrotoxicose en raison du renouvellement accru de PIP2 et les canaux Kir interagissent directement avec PIP2 pendant le déclenchement normal. Dans le syndrome d’Andersen-Tawil, il y a une diminution de l’affinité PIP2. Dans la PP thyrotoxique, aucune des mutations ne modifie la rectification de Kir2.6.