Rapport de cas de gynécologie et d’obstétrique
Mots clés
Tetra-amelia; Malformation; Génotype; Phénotype.
Introduction
Les anomalies des membres constituent un groupe important de pathologies congénitales caractérisées par une hypoplasie ou une aplasie complète d’un ou plusieurs os des membres. Des anomalies des membres de tous types surviennent dans environ 1 naissance sur 1 300 à 2 000. Ces anomalies des membres peuvent être isolées ou associées à d’autres anomalies. Le syndrome de tétraamélie est rare et des zones grises subsistent.
Nous rapportons deux cas de tétraamélie dans une maternité de niveau II à Dakar (Sénégal) avec pour être similaire à la tétraamélie-1 (chromosome 17q21), la tétraamélie-2 (chromosome 8q23) et le syndrome de Robert (chromosome 8p21). Cela illustre la difficulté de corréler le phénotype et les gènes impliqués.
Rapports de cas
Cas 1
Mme AD était une mère de 44 ans référée à notre service à 36 semaines de gestation avec une pré-éclampsie sévère et des anomalies fœtales. Elle avait cinq ans et n’avait aucun antécédent d’anomalies fœtales. Elle ne fumait pas maintenant et n’avait jamais ni fumé ni bu d’alcool. Elle n’avait pas été exposée au tabagisme passif. Elle était dans un mariage consanguin au troisième degré pour tous ses enfants. Mme AD avait des tests négatifs pour l’hépatite B, le VIH et la syphilis. Elle était protégée contre le virus de la rubéole et n’avait aucune exposition antérieure à Toxoplasma gondii. La surveillance échographique effectuée tardivement à 33 semaines et 35 semaines de gestation a permis de retrouver une oligoamniose et une hydrocéphalie ainsi qu’une agénésie des membres. Les prescriptions pendant la grossesse comprenaient l’administration de fer et d’acide folique, ainsi que l’administration de sulfadoxine pyriméthamine. Ce dernier a été prescrit à 18 semaines puis 26 semaines dans le cadre de la politique de prophylaxie antipaludique pour les femmes enceintes. La hauteur symphysaire-fondamentale mesurait 28 cm. En raison de caractéristiques sévères de pré-éclampsie; elle a été hospitalisée immédiatement et observée dans une unité de travail et d’accouchement. Elle a ensuite d’abord reçu du sulfate de magnésium par voie intraveineuse pour prévenir l’éclampsie et des médicaments antihypertenseurs pour maintenir la pression artérielle systolique en dessous de 160 mmHg et la pression artérielle diastolique en dessous de 105 mmHg.
La décision d’un accouchement par césarienne immédiat a été prise. Un vivant de 2 150 grammes a été extrait qui est ensuite mort en 10 minutes. Avant la mort, le corps présentait des mouvements rampants. Plusieurs anomalies externes ont été identifiées (Figure 1), notamment une agénésie complète des quatre membres, une hydrocéphalie avec un tour de tête de 39 cm. Sur le visage, il y avait un hypertélorisme avec un colobome des paupières, une exophtalmie légère et une aniridie. La bouche était semblable à un V inversé sans délimitation claire des lèvres et le nez était rudimentaire. Le nouveau-né était dépourvu de téguments (cheveux et sourcils). Les oreilles étaient réduites à des croquis qui ressemblaient à des fentes. Le cou était court. Le tronc était réduit à une structure conique de 26 cm de long avec un cordon ombilical à l’extrémité inférieure. La poitrine était étroite. Juste en dessous du nombril, le tronc élargi vers l’arrière était présent au niveau du plancher du pôle caudal, une ligne médiane avec des récessions et un bourgeonnement pouvant correspondre à un phallus de type indéterminé. Une agénésie du bassin, des organes génitaux et une imperforation anale ont été notées. La pathologie fœtale n’a pas été réalisée. Cependant, la mort dans les 10 minutes suivant l’accouchement et l’aspect conique de la poitrine peuvent suggérer des anomalies pulmonaires.
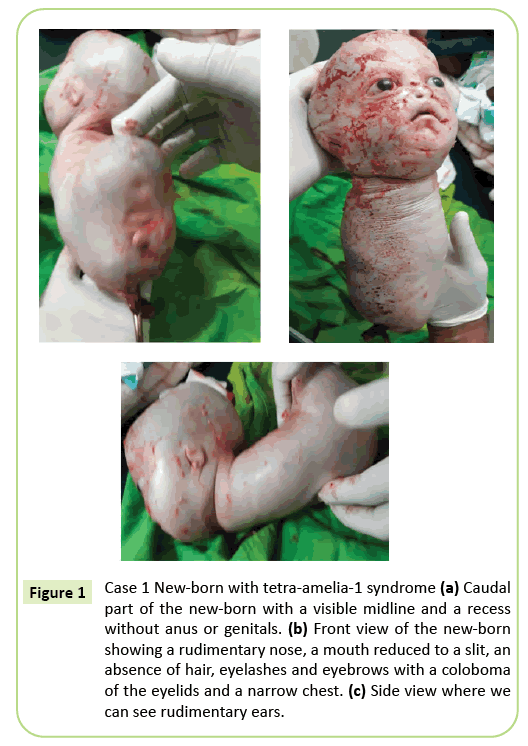
Figure 1: Cas 1 Nouveau-né avec syndrome de tetra-amelia-1 (a) Partie caudale du nouveau-né avec une ligne médiane visible et un renfoncement sans anus ni organes génitaux. b) Vue de face du nouveau-né montrant un nez rudimentaire, une bouche réduite à une fente, une absence de poils, de cils et de sourcils avec un colobome des paupières et une poitrine étroite. (c) Vue de côté où l’on peut voir des oreilles rudimentaires.
Cas 2
Le deuxième cas était un primigravida âgé de 22 ans renvoyé à notre établissement pour une échographie à 37 semaines de gestation. Elle n’était pas dans un mariage consanguin. Elle avait été testée négative pour l’hépatite B, le VIH et la syphilis. Elle n’a pas été testée pour la toxoplasmose et la rubéole. Aucune surveillance échographique n’a été effectuée pendant sa grossesse. L’examen clinique a été cohérent avec un retard de croissance fœtale (hauteur du fond: 26 cm). Les résultats échographiques ont montré que l’humérus était déformé en mesurant 23,9 mm correspondant à 17 semaines de gestation. Il y avait une agénésie du fémur. Les ailes iliaques étaient visibles à l’échographie. Aucune anomalie pulmonaire ou cardiaque n’a été identifiée. La livraison a été lancée. Le nouveau-né avait un phénotype femelle avec un score Apgar de 9 à la 5e minute. La morphologie de la tête et du tronc était sans particularité. Les membres supérieurs ont été réduits à deux souches de 3 cm de long. Une agénésie complète des 2 membres inférieurs a été notée. C’était une anomalie symétrique (figure 2).
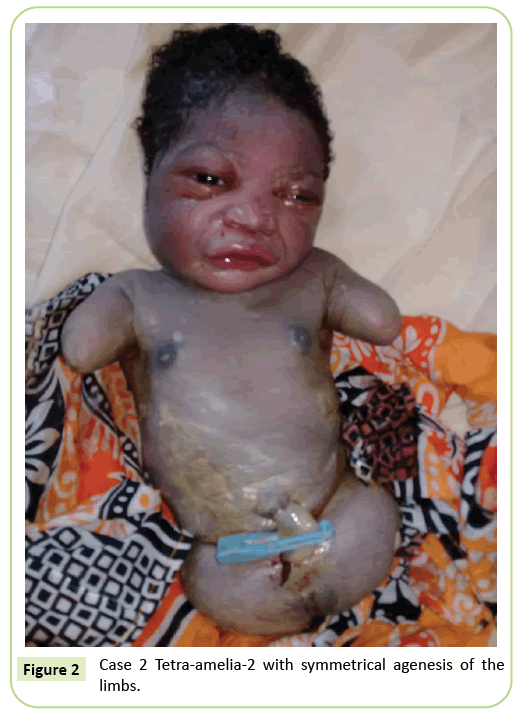
Figure 2: Cas 2 Tétra-amélia-2 avec agénésie symétrique des membres.
Discussion
Bermejo-Sanchez et coll. a décrit en 2011 l’épidémiologie de l’amélia congénitale à l’aide de données collectées à partir de 20 programmes de surveillance des anomalies congénitales, de tous les continents sauf l’Afrique, entre 1968 et 2006. Au total, 326 cas d’amelia ont été identifiés parmi 23 110 591 naissances vivantes, mortinaissances et avortements. La prévalence était de 1,41 / 100 000.
Tetra-amelia se réfère à l’absence complète des membres et se produit plus rarement. À notre connaissance, tetra-amelia-1 est décrit en 7 familles. Il semble suivre un héritage autosomique récessif. Dans toutes les familles, tetra-amelia-1 était associée à des malformations graves des autres parties du corps, y compris le visage et la tête, des anomalies du système nerveux, du squelette et des organes génitaux. Dans de nombreux cas, les poumons étaient sous-développés, ce qui rendait la respiration difficile ou impossible. Zimmer et coll. signalé en 1985 une famille fortement consanguine dans laquelle 6 nourrissons avaient une tétra-amélia-1 et une hydrocéphalie. Ils ont décrit chez l’un des fœtus une absence totale d’os pelvien, une fente labiale et palatine, une arrhinie et une aplasie des oreilles. Un poumon gauche bilatéral, un canal artériel persistant, une imperforation anale ont également été notés. Les tests fœtaux ont éliminé le diagnostic du syndrome de Robert. D’autres cas retrouvés dans la littérature comprennent celui de Kosaki et coll., en 1996, avec un fœtus de caryotype 46, XX avec tétraphocomélie et hypoplasie pulmonaire sévère en plus d’anomalies du visage et de la tête. Rosenak et coll. décrit un cas de tétra-amélia avec hypoplasie pulmonaire sévère chez deux fœtus d’un couple non consanguin. Les tests fœtaux ont exclu le diagnostic du syndrome de Robert. Deux autres cas ont été signalés par Zlotogora et al. en 1993. Les deux patients sont décédés peu de temps après la naissance et les auteurs ont suggéré l’existence d’une hypoplasie pulmonaire. Niemann et coll. a rapporté une famille turque consanguine dans laquelle 4 des 8 frères souffraient de tétra-amélia. En plus de l’absence des 4 membres, les examens fœtaux de 3 fœtus ont révélé de multiples anomalies: fente labiale et / ou palatine, laparoschisis, anomalies pulmonaires, hypoplasie du bassin, atrésie des choanas, imperforation vaginale et anale. Enfin, en 2005, Krahn et al. décrit 2 frères nés de parents consanguins souffrant de tétraamélie et d’hypoplasie pulmonaire sévère. Les clavicules et les omoplates étaient normales chez le deuxième fœtus. Le caryotype était normal.
Le syndrome de Tétra-amélia-1 ou TÉTAMS1 est causé par une mutation homozygote du gène WNT3 sur le chromosome 17q21 avec un héritage autosomique récessif. Le syndrome de tétraamélie-2 (TÉTAMS2) est caractérisé par des membres rudimentaires ou une absence complète des membres, généralement symétriques ainsi qu’une agénésie bilatérale des poumons dans certains cas. Sont également des anomalies habituelles du système vasculaire pulmonaire et des dysmorphies, notamment une fente labiale et palatine bilatérale, une ankyloglossie, une hypoplasie mandibulaire, une microrétrognathie et une aplasie labioscrotale.
Szenker-Ravi, en étudiant 4 familles de tétra-amélia avec agénésie ou hypoplasie pulmonaire, a noté une hétérogénéité phénotypique avec des anomalies des membres de gravité variable. Le séquençage d’exomes dans ces 4 familles a permis d’identifier des mutations homozygotes tronquées dans le gène RSPO2. Le syndrome de tétraamélie-2 est causé par une mutation homozygote du gène RSPO2 (610575) situé sur le chromosome 8q23.
Le phénotype du premier cas décrit dans cet article correspond à un syndrome tetra-amelia-1 dû notamment à la présence d’hydrocéphalie, d’anomalies des organes génitaux et d’un nez rudimentaire. La poitrine étroite et la mort précoce avant la 10e minute de vie suggèrent une hypoplasie pulmonaire sévère. Ce cas met en évidence l’hétérogénéité phénotypique avec un colobome des paupières, un hypertélorisme, une exophtalmie et des appendices rares.
Nous considérons que le deuxième cas de notre étude est un syndrome de tétramélie-2 compte tenu de la tétramélie symétrique avec présence de souches de membres supérieurs. Le diagnostic de tétra-amélia doit être effectué tôt lors de la surveillance échographique. Par conséquent, il convient de sensibiliser à l’importance de la surveillance échographique et de l’utilisation de la 3D / 4D pour améliorer les résultats du dépistage. Le diagnostic d’une masse pelvienne à l’échographie associée à amelia devrait éveiller la suspicion de syndrome de défaut des membres de fusion spléno-gonadique.
De plus, l’examen fœtal et les tests fœtaux utilisant les technologies évolutives de la microréseau chromosomique et du séquençage des exomes et du génome doivent être encouragés dans nos contextes. Une meilleure caractérisation des cas permet d’apporter des conseils aux couples et une meilleure connaissance de ces anomalies cliniques.
Conclusion
Le syndrome de tétra-amélia est rare et des zones grises subsistent. Ces deux cas, comparés à ce qui est déjà décrit dans la littérature, illustrent l’hétérogénéité phénotypique de la tétraamélie. Compte tenu de la fréquence rare de ces anomalies, il serait important de créer un registre international des anomalies afin de signaler les cas et de créer une banque d’échantillons pour des études génétiques étendues aux parents.
- Wilcox WR, Coulter CP, Schmitz ML (2015) Troubles de déficience congénitale des membres. Clin Périnatol 42:281-300.
- Bermejo SE, Cuevas L, Amar E (2011) Amelia: Une étude épidémiologique descriptive multicentrique dans un vaste ensemble de données de l’International Clearinghouse for Birth Defects Surveillance and Research, et vue d’ensemble de la littérature. Am J Med Genet C Semin Med Genet 157:288-304.
- Zlotogora JSM, Shabany YO, Jarallah RY (1993) Syndrome de tétraamélie avec hypoplasie pulmonaire. Am J Med Genet 47:570-571.
- Zimmer EZ (1985) Tetra-amélia avec malformations multiples chez six fœtus mâles d’une même famille. Europ. J Pediat 144:412-414.
- Rosenak D (1991) Tétraamélie récurrente et hypoplasie pulmonaire avec malformations multiples chez les frères et sœurs. Am J Med Genet 38, 25-28.
- Syndrome de Gershoni BR (1990) Roberts ou « amelia liée à l’X »? . Am J Med Genet 37:569-572.
- Kosaki K, Jones MC, Stayboldt C (1996) Zimmer phocomelia: délimitation par analyse des coordonnées principales. Am J Med Genet 66, 55-59.
- La mutation homozygote WNT3 de Niemann S(2004) provoque une tétra-amélia dans une grande famille consanguine. Am J Hum Genet 74:558-563.
- Krahn M (2005) Tetra-amelia et syndrome d’aplasie pulmonaire: rapport d’une nouvelle famille et exclusion de gènes candidats. Clin Genet 68:558-560.
- Szenker-RE, Altunoglu U (2018) L’inhibition de RSPO2 du RNF43 et du ZNRF3 régit le développement des membres indépendamment du LGR4/5/6. Nature 557:564-569.