Syndrome d’Aicardi-Goutières: Spectre phénotypique et génétique dans une série de trois cas / Anales de Pediatría
Le syndrome d’Aicardi-Goutières (AGS) est une maladie héréditaire rare dont la prévalence exacte est inconnue. Elle a été décrite pour la première fois en 1984 par Jean Aicardi et Françoise Goutières comme une encéphalopathie progressive avec apparition dans les premiers mois de la vie caractérisée par une lymphocytose du liquide céphalo-rachidien (LCR) et des calcifications dans les ganglions de la base.1 Il se manifeste par une irritabilité, un retard psychomoteur, une spasticité, une dystonie, des crises d’épilepsie, des épisodes récurrents de fièvre aseptique et de microcéphalie. La mortalité est plus élevée pendant la phase encéphalopathique, et bien que la maladie se stabilise généralement après, elle provoque de graves séquelles neurologiques. D’autres caractéristiques qui peuvent apparaître au cours de son évolution sont les engelures, les symptômes oculaires (principalement le glaucome), l’atteinte cardiaque ou les troubles auto-immunes.2 Les interférons de type I jouent un rôle crucial dans la pathogenèse de l’AGS, dans laquelle leur expression est régulée à la hausse, ce qui entraîne une augmentation de la production.3 Pour cette raison, l’un des résultats de laboratoire classiques chez ces patients est un taux élevé d’interféron alfa dans le LCR, ainsi qu’une pléocytose et des taux tout aussi élevés de néoptérine et de bioptérine. L’utilité potentielle d’évaluer le niveau d’expression des gènes stimulés par l’interféron dans le sang périphérique en tant que marqueur est actuellement à l’étude, car il existe des preuves que ces niveaux restent élevés au-delà de la phase encéphalopathique (« signature interféron »).3-5 Une autre caractéristique clé est la détection d’anomalies de neuroimagerie, y compris des calcifications dans les ganglions de la base et des modifications de la substance blanche (Fig. 1). À ce jour, nous connaissons 7 gènes dont les mutations peuvent conduire à une régulation à la hausse de la voie de l’interféron : ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1 et IFIH1. Des mutations hétérozygotes ont été décrites pour les gènes TREX1, ADAR et IFIH1, alors que les mutations rapportées dans tous les autres gènes ont été homozygotes.2 Les mutations du gène IFIH1 ont été détectées le plus récemment (2014)4 et sont donc les variants pathogènes les moins fréquents, alors que les mutations des gènes RNASEH2B et TREX1 représentent la plus forte proportion de cas diagnostiqués d’AGS.
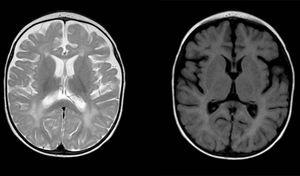
Anomalies diffuses et parcellaires du signal dans la substance blanche dans les deux hémisphères cérébraux, hyperintense dans les images pondérées en T2. Espace sous-arachnoïdien élargi avec prédominance frontotemporale dans les deux hémisphères, avec élargissement de la fissure interhémisphérique et augmentation de la taille ventriculaire (en l’absence d’augmentation de la pression), compatible avec l’atrophie corticale et sous-corticale.
Au cours des dernières décennies, grâce aux progrès de la génétique permettant la détection de ces mutations spécifiques, des preuves ont émergé d’un large spectre phénotypique au-delà de la présentation classique basée sur le gène responsable. Nous présentons les cas de 3 patients ayant reçu un diagnostic d’AGS au cours des 8 dernières années dans le but d’analyser leurs caractéristiques cliniques par rapport au défaut génétique sous-jacent (tableau 1). En général, les caractéristiques de présentation de l’AGS étaient cohérentes avec celles décrites dans la série de cas la plus récente dans la littérature: présentation néonatale (33%), microcéphalie (66%), retard psychomoteur (100%), spasticité (100%), déficience intellectuelle sévère (66%) et calcifications sur la tomodensitométrie crânienne (66%), bien qu’un seul patient ait eu des crises d’épilepsie.
Caractéristiques des patients atteints du syndrome d’Aicardi-Goutières.
| Affaire 1 | Affaire 2 | Affaire 3 | |
|---|---|---|---|
| Génétique | Mutation homozygote (p. Ala177Thr) dans le gène RNASEH2B | Mutation homozygote (341G >A) dans le gène TREX1 | Mutation hétérozygote (c. 992C > G et p.Thr331Arg) dans le gène IFIH1 |
| Âge actuel | 3 ans | 7 ans et 4 mois | 12 ans et 11 mois |
| Sexe | Mâle | Femelle | Mâle |
| Origine | Roumanie | Espagne | Italie |
| AP | – | Semaine 36: restriction de croissance intra-utérine Semaine 37: microcéphalie, calcification placentaire |
Fente palatine |
| Manifestations cliniques | |||
| Âge au début | 10 mois | Naissance | 2 ans |
| Présentation initiale | Irritabilité Régression psychomotrice |
Tremblements, hypotonie, pleurs faibles, échec de la croissance | Retard de motricité |
| Retard psychomoteur | Oui | Oui | Oui |
| Langue | 2 syllabes words | No | Yes |
| Microcephaly | Yes | Yes | No |
| Chilblains | No | Yes | Yes |
| Epilepsy | No | Yes | No |
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFCS IV |
| Trouble du mouvement | Non | Oui | Non |
| Mouvements oculaires anormaux | Non | Non | Non |
| Déficience visuelle | Non | – | Myopie |
| Glaucome | Non | Non | Non |
| Perte auditive | – | – | Non |
| Atteinte cardiaque | Non | Régurgitation tricuspide et mitrale légère | Non |
| Fièvre récurrente | Non | Non | Non |
| Déficience intellectuelle | Oui | Oui, grave | Oui, légère |
| Autres | – | – | Syndrome de Singleton-Merten: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
| CSF | |||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – |
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – |
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – |
| INF alfa (IU/mL) | 25 ( | 75 ( | – |
| Blood | |||
| Interferon signature | – | – | Gene overexpression |
| Neuroimaging | |||
| Calcifications on CT | No | Yes. Dans les ganglions basaux et périventriculaires | Oui. Calcifications symétriques dans la MW profonde de la région frontale et du noyau lentiforme |
| IRM de la tête | Changements diffus et inégaux de l’intensité de la MW des deux hémisphères cérébraux, hyperintense sur T2. Atteinte de la MW sous-corticale (épargnant les fibres U) et de la MW périventriculaire | Atteinte généralisée de la MW avec prédominance de la MW lobaire, y compris les fibres U sous-corticales des lobes frontal, temporal et occipital, bilatéralement et symétriquement, sans atteinte corticale | – |
LCR, liquide céphalo-rachidien; TDM, tomodensitométrie; GMFCS, Système de classification de la Fonction Motrice Brute; INF, interféron; RCIU, restriction de croissance intra-utérine; IRM, imagerie par résonance magnétique; PNP, polyneuropathie; WM, mater blanche.
Comme indiqué précédemment, les mutations homozygotes dans le gène RNASEH2B sont les variantes les plus fréquentes qui causent l’AGS et leur expression phénotypique est généralement la plus conforme à la présentation classique.4 C’était le cas du patient de notre étude porteur d’une telle mutation, qui était apparu à l’âge de 10 mois avec irritabilité et retard psychomoteur et avec des résultats de neuroimagerie et de LCR caractéristiques.
Vingt pour cent des cas d’AGS peuvent avoir une présentation néonatale, avec l’apparition de la maladie in utero.5 Mutations dans l’un des 7 gènes susmentionnés peuvent conduire à ce phénotype, mais cette présentation précoce est le plus souvent associée au gène TREX.4,5 La présentation initiale de cette forme est similaire à celle d’une infection TORCH, avec hépatosplénomégalie, hypertransaminasémie, thrombocytopénie et manifestations neurologiques comprenant une irritabilité extrême, des troubles du mouvement et des crises d’épilepsie.5 Ces patients ont une évolution plus sévère de la maladie et courent un risque plus élevé de décès. Le patient de notre échantillon qui présentait une telle variante avait une présentation néonatale et a actuellement la forme de maladie la plus grave du 3.
Les mutations du gène ADAR1 et surtout du gène IFIH1 sont associées à une apparition tardive des symptômes, après 1 an de vie avec un développement psychomoteur normal.5 Dans certains de ces cas, le syndrome a une évolution bénigne avec une préservation relative du langage et de la motricité. Notre patient avec une mutation du gène IFIH1 était un cas singulier en ce sens qu’il était également atteint du syndrome de Singleton-Merten, une maladie rare également causée par une mutation du gène IFIH1 et caractérisée par une dysplasie dentaire, des calcifications aortiques et de l’ostéoporose.6
Notre objectif est de souligner la variabilité phénotypique significative de l’AGS et son association avec des mutations spécifiques dans le but à la fois d’encourager la prise en compte de ce diagnostic dans les cas avec des présentations qui s’écartent de la forme classique de la maladie et d’apporter des informations supplémentaires sur l’évolution de la maladie et les résultats chez ces patients.