Gynækologi & Obstetrik Case report
nøgleord
Tetra-amelia; misdannelse; Genotype; fænotype.
introduktion
Limb anomalier udgør en vigtig gruppe af medfødte patologier karakteriseret ved hypoplasi eller fuldstændig aplasi af en eller flere lemmer knogler. Lemabnormiteter af alle typer forekommer hos cirka 1 ud af 1.300 til 2.000 fødsler. Disse lemanomalier kan isoleres eller forbindes med andre anomalier . Tetraamelia syndrom er sjældent, og grå områder forbliver.
vi rapporterer to tilfælde af tetra-amelia i et niveau II-moderskab i Dakar (Senegal) med at ligne tetraamelia-1 (kromosom 17k21), tetraamelia-2 (kromosom 8k23) og Roberts syndrom (kromosom 8p21). Dette illustrerer vanskeligheden ved at korrelere fænotype og involverede gener.
Sagsrapporter
sag 1
Ms. AD var en 44-årig mor henvist til vores afdeling ved 36 ugers drægtighed med svær præeklampsi og føtale anomalier. Hun var fem para uden historie med føtale anomalier. Hun røg ikke nu og havde aldrig hverken røget eller drukket alkohol. Hun havde ikke været udsat for passiv rygning. Hun var i et tredje grads sammenhængende ægteskab for alle sine børn. MS AD havde testet negativt for hepatitis B, HIV og syfilis. Hun var beskyttet mod røde hundevirus og havde ingen tidligere eksponering for toksoplasma gondii. Ultralyd overvågning udført sent på 33 uger og 35 ugers svangerskab hentet oligoamniosis og hydrocephalus samt agenese af lemmerne. Recept under graviditet omfattede administration af jern og folsyre samt administration af sulfadoksin pyrimethamin. Sidstnævnte blev ordineret efter 18 uger og derefter 26 uger som en del af anti-malaria-profylaksepolitikken for gravide kvinder. Symphyseal-fundal højde målt 28 cm. På grund af alvorlige træk ved præeklampsi; hun blev straks indlagt på hospitalet og observeret i en arbejds-og leveringsenhed. Hun modtog derefter oprindeligt IV magnesiumsulfat for at forhindre eklampsi og antihypertensive medicin for at opretholde systolisk blodtryk under 160 mmHg og diastolisk blodtryk under 105 mmHg.
beslutningen om øjeblikkelig kejsersnit blev truffet. En 2.150 gram levende født blev ekstraheret, som efterfølgende døde inden for 10 minutter. Før døden præsenterede kroppen krybende bevægelser. Flere eksterne anomalier blev identificeret (Figur 1) inklusive komplet agenese af alle fire lemmer, hydrocephalus med en hovedomkreds på 39 cm. På ansigtet var der hypertelorisme med øjenlågens colobom, mild eksophthalmos og aniridia. Munden var ens en omvendt V uden klar afgrænsning af læberne og næsen var rudimentær. Den nyfødte var uden integumenter (hår og øjenbryn). Ørene blev reduceret til skitser, der lignede slidser. Halsen var kort. Bagagerummet blev reduceret til en 26 cm lang konisk struktur med en navlestreng i bundenden. Brystet var smalt. Lige under navlen var bagagerummet udvidet bagud til stede på gulvet i den kaudale Pol, en midtlinie med recessioner og en spirende, der kan svare til en fallus af ubestemt type. Agenese af bækkenet, kønsorganer og anal ufuldkommenhed blev noteret. Fosterpatologi er ikke udført. Imidlertid kan døden inden for 10 minutter efter fødslen og det koniske udseende af brystet antyde lungeabnormiteter.
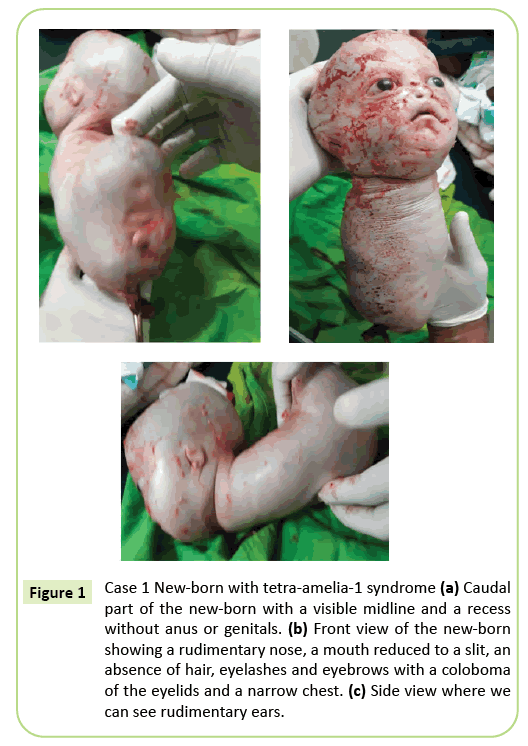
Figur 1: Sag 1 nyfødt med tetra-amelia-1 syndrom (a) Caudal del af den nyfødte med en synlig midtlinie og en fordybning uden anus eller kønsorganer. (B) set forfra af den nyfødte, der viser en rudimentær næse, en mund reduceret til en spalte, fravær af hår, øjenvipper og øjenbryn med øjenlågens colobom og et smalt bryst. (C) set fra siden, hvor vi kan se rudimentære ører.
sag 2
den anden sag var en 22-årig primigravida henvist til vores facilitet til ultralydsscanning ved 37 ugers drægtighed. Hun var ikke i et sammenhængende ægteskab. Hun havde testet negativt for hepatitis B, HIV og syfilis. Hun blev ikke testet for toksoplasmose og røde hunde. Der blev ikke foretaget ultralydsovervågning under hendes graviditet. Den kliniske undersøgelse var i overensstemmelse med føtal væksthæmning (Fundal højde: 26 cm). Ultralydsfund viste, at humerus var forvrænget, der måler 23,9 mm svarende til 17 ugers drægtighed. Der var agenese af lårbenet. Iliac vingerne var synlige på ultralyd. Ingen lunge-eller hjerteabnormiteter blev identificeret. Levering blev indledt. Den nyfødte havde en kvindelig fænotype med en Apgar-score på 9 i 5.minut. Morfologien på hovedet og bagagerummet var uden særlig karakter. De øvre lemmer blev reduceret til to 3 cm lange stubbe. Komplet agenese af de 2 nedre lemmer blev noteret. Det var en symmetrisk anomali (figur 2).
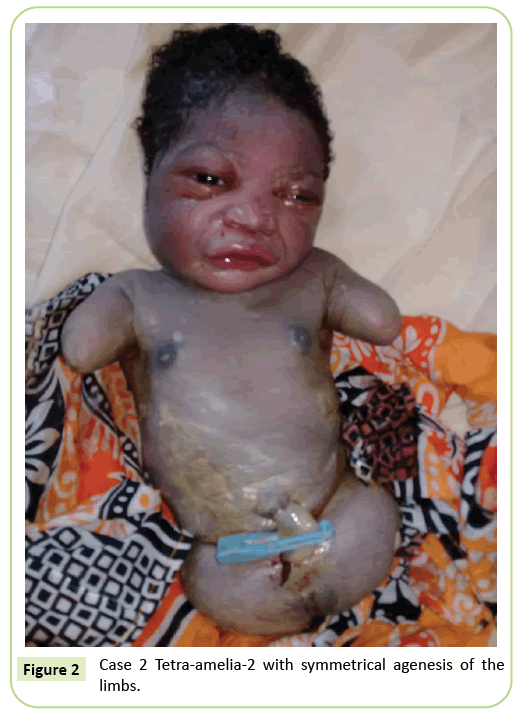
figur 2: sag 2 Tetra-amelia-2 med symmetrisk agenese af lemmerne.
Diskussion
Bermejo-Sanches et al. beskrevet i 2011 epidemiologien af medfødt amelia ved hjælp af data indsamlet fra 20 medfødte anomalier overvågningsprogrammer fra alle kontinenter undtagen Afrika mellem 1968 og 2006. I alt blev 326 tilfælde af amelia identificeret blandt 23.110.591 levende fødsler, dødfødsler og aborter. Prævalensen var 1,41 / 100.000 .
Tetra-amelia refererer til som fuldstændig fravær af lemmerne og forekommer sjældnere. Så vidt vi ved, er tetra-amelia-1 beskrevet i 7 familier. Det ser ud til at følge en autosomal recessiv arv. I alle familier var tetra-amelia-1 forbundet med alvorlige misdannelser i de andre dele af kroppen, herunder ansigt og hoved, anomalier i nervesystemet, skelet og kønsorganer. Lungerne var underudviklede i mange tilfælde, hvilket gør vejrtrækning vanskelig eller umulig . Et al. rapporteret i 1985 en stærkt indavlet familie, hvor 6 spædbørn havde tetra-amelia-1 og hydrocephalus. De beskrev i et af fostrene et totalt fravær af bækkenbenet, spaltet læbe og gane, arrhinia og aplasi i ørerne. En bilateral venstre lunge, en vedvarende arteriel kanal, en anal ufuldkommenhed blev også bemærket. Fostertest eliminerede diagnosen Roberts syndrom . Andre sager hentet i litteraturen inkluderer Kosaki et al., i 1996, med et foster af karyotype 46, KS med tetraphocomelia og svær lungehypoplasi ud over ansigts-og hovedanomalier . Rosenak et al. beskrevet et tilfælde af tetra-amelia med svær lungehypoplasi hos to fostre af et ikke-sammenhængende par. Fostertest udelukkede diagnose af Roberts syndrom . To yderligere tilfælde blev rapporteret af Llotogora et al. i 1993. Begge patienter døde kort efter fødslen, og forfatterne foreslog eksistensen af lungehypoplasi. Niemann et al. rapporterede en sammenhængende tyrkisk familie, hvor 4 af de 8 brødre led af tetra-amelia. Ud over fraværet af de 4 lemmer afslørede føtalundersøgelserne af 3 fostre flere anomalier: spaltede læber og /eller palatin, laparoschisis, lungeanomalier, bækkenets hypoplasi, atresi af choanas, vagina og anal imperforation . Endelig, i 2005, Krahn et al. beskrevet 2 brødre født til indavlede forældre, der lider af tetraamelia og svær lungehypoplasi. Kraveben og skulderblade var normale i det andet foster. Karyotypen var normal .
Tetra-amelia-1 syndrom eller TETAMS1 er forårsaget af en homosygøs mutation i VNT3-genet på kromosom 17k21 med en autosomal recessiv arv. Tetraamelia-2 syndrom (TETAMS2) er kendetegnet ved rudimentære lemmer eller et fuldstændigt fravær af lemmerne, generelt symmetrisk såvel som bilateral agenese i lungerne i nogle tilfælde. Er også sædvanlige anomalier i det pulmonale vaskulære system og dysmorfier, herunder bilateral læbe-og ganespalte, ankyloglossi, mandibulær hypoplasi, mikroretrognathia og labioscrotal aplasi .
Ssenker-Ravi, der studerede 4 familier af tetra-amelia med agenese eller pulmonal hypoplasi, bemærkede en fænotypisk heterogenitet med lemanomalier af forskellig sværhedsgrad . Eksomsekventering i disse 4 familier har gjort det muligt at identificere trunkerende homosygøse mutationer i RSPO2-genet . Tetraamelia-2 syndrom er forårsaget af en homosygøs mutation i rspo2-genet (610575) placeret på kromosom 8k23 .
fænotypen af det første tilfælde beskrevet i denne artikel svarer til et tetra-amelia-1-syndrom, der især skyldes tilstedeværelsen af hydrocephalus, anomalier kønsorganer og en rudimentær næse. Det smalle bryst og den tidlige død inden det 10.minut af livet antyder alvorlig lungehypoplasi. Denne sag fremhæver den fænotypiske heterogenitet med et øjenlågskolobom, hypertelorisme, eksophthalmos og sjældne vedhæng.
vi betragter det andet tilfælde i vores undersøgelse som et tetramelia-2-syndrom i betragtning af den symmetriske tetra-amelia med tilstedeværelsen af stubbe i øvre lemmer. Diagnose af tetra-amelia skal udføres tidligt under ultralydsovervågning. Derfor bør man være opmærksom på vigtigheden af ultralydsovervågning og brugen af 3d/4d for at forbedre screeningsresultaterne. Diagnosen af en bækkenmasse ved ultralyd parret med amelia bør rejse mistanke om spleno-gonadal fusion limb defect syndrome.
derudover skal fosterundersøgelsen og fostertest ved hjælp af de udviklende teknologier for kromosomal mikroarray og eksom-og genomsekventering opmuntres i vores indstillinger. En bedre karakterisering af sagerne gør det muligt at rådgive par og et bedre kendskab til disse kliniske anomalier.
konklusion
Tetra-amelia syndrom er knappe, og der er stadig grå områder. Disse to tilfælde sammenlignet med det, der allerede er beskrevet i litteraturen, illustrerer tetraameliens fænotypiske heterogenitet. I betragtning af den sjældne forekomst af disse anomalier ville det være vigtigt at oprette et internationalt register over anomalier for at rapportere sager og oprette en prøvebank til udvidede genetiske undersøgelser til forældre.
- Congenital lem deficiency disorders (2015). Clin Perinatol 42: 281-300.
- Bermejo SE, Cuevas L, Amar E (2011) Amelia: en multi-center beskrivende epidemiologisk undersøgelse i et stort datasæt fra International Clearinghouse for Fødselsdefektovervågning og forskning og oversigt over litteraturen. Am J Med Genet C Semin Med Genet 157: 288-304.
- tslotogora JSM, Shabany YO, Jarallah RY (1993) syndrom af tetraamelia med lungehypoplasi. Am J Med Genet 47: 570-571.
- simmer es (1985) Tetra-amelia med flere misdannelser hos seks mandlige fostre i en slægt. Europ. J Pediat 144: 412-414.
- Rosenak D (1991) tilbagevendende tetraamelia og pulmonal hypoplasi med multiple misdannelser hos SIB ‘ er. Am J Med Genet 38: 25-28.
- Gershoni BR (1990) Roberts syndrom eller “Amelia” ? . Am J Med Genet 37: 569-572.
- Kosaki K, Jones MC, Stayboldt C (1996) simmer phocomelia: afgrænsning ved hovedkoordinatanalyse. Am J Med Genet 66: 55-59.
- Niemann S(2004) homosygøs vnt3-mutation forårsager tetra-amelia i en stor sammenhængende familie. Am J Hum Genet 74: 558-563.
- Krahn M (2005)Tetra-amelia and lung aplasia syndrome: rapport om en ny familie og udelukkelse af kandidatgener. Clin Genet 68: 558-560.
- Ssenker-RE, Altunoglu U (2018) rspo2-hæmning af RNF43 og RNRF3 styrer udviklingen af lemmer uafhængigt af LGR4/5/6. Natur 557: 564-569.