Aicardi-Gouti GmbH-szindróma: fenotípusos és genetikai spektrum három esetből álló sorozatban / Anales de Pediatr adapta
az Aicardi-Gouti GmbH-szindróma (AGS) ritka örökletes betegség, amelynek pontos előfordulása nem ismert. Először 1984-ben írta le Jean Aicardi és Francoise Gouti, mint progresszív encephalopathia, amely az élet első hónapjaiban jelentkezik, melyet cerebrospinális folyadék (CSF) limfocitózis és a bazális ganglionok meszesedése jellemez.1 az ingerlékenység, a pszichomotoros retardáció, a spaszticitás, a dystonia, az epilepsziás rohamok, az aszeptikus láz és a mikrocefália visszatérő epizódjai jelentkeznek. A mortalitás magasabb az encephalopathiás fázisban, és bár a betegség jellemzően később stabilizálódik, súlyos neurológiai következményeket okoz. Egyéb jellemző tulajdonságok, amelyek a tanfolyam során megjelenhetnek, a chilblains, a szem tünetei (főleg glaukóma), a szív érintettsége vagy autoimmun rendellenességek.2 Az I. típusú interferonok döntő szerepet játszanak az AGS patogenezisében, amelyben expressziójuk szabályozva van, ami fokozott termeléshez vezet.3 ezért az egyik klasszikus laboratóriumi lelet ezeknél a betegeknél az alfa-interferon emelkedett szintje a CSF-ben, a pleocytosissal együtt, valamint a neopterin és a biopterin egyaránt emelkedett szintje. Az interferon által stimulált gének interferon általi expressziójának markerként történő értékelésének potenciális hasznosságát jelenleg vizsgálják, mivel bizonyítékok vannak arra, hogy ezek a szintek magasan maradnak az encephalopathiás fázison túl (“interferon aláírás”).3-5 egy másik kulcsfontosságú jellemző a neuroimaging rendellenességek kimutatása, beleértve a bazális ganglionok meszesedését és a fehérállomány változásait (ábra. 1). Eddig 7 olyan génről tudunk, amelyek mutációi az interferon útvonalának szabályozásához vezethetnek: ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1 és IFIH1. Heterozigóta mutációkat írtak le a TREX1, ADAR és IFIH1 gének esetében, míg az összes többi génben jelentett mutációk homozigóták voltak.2 az IFIH1 gén mutációit legutóbb (2014)4 detektálták, ezért ezek a legkevésbé gyakori patogén variánsok, míg az RNASEH2B és a TREX1 gének mutációi teszik ki az AGS diagnosztizált eseteinek legnagyobb arányát.
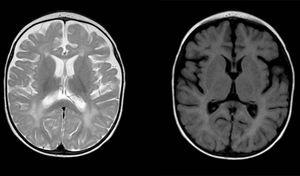
diffúz és foltos jel rendellenességek a fehérállományban mindkét agyféltekében, hiperintenzitás a T2-súlyozott képeken. Megnagyobbodott subarachnoidális térben frontotemporal túlsúlya mindkét féltekén, a kiszélesedő interhemisphericus hasadék és a megnövekedett kamrai méret (hiányában a megnövekedett nyomás), kompatibilis kortikális és subcorticalis atrophia.
az elmúlt néhány évtizedben, a genetika fejlődésének köszönhetően, amely lehetővé tette ezen specifikus mutációk kimutatását, bizonyítékok merültek fel egy széles fenotípusos spektrumról, amely túlmutat a kórokozó génen alapuló klasszikus bemutatáson. Bemutatjuk 3 beteg eseteit, akiknél az elmúlt 8 évben diagnosztizálták az AGS-t azzal a céllal, hogy elemezzék klinikai jellemzőiket a mögöttes genetikai hibához viszonyítva (1.táblázat). Általánosságban elmondható, hogy az AGS megjelenési jellemzői összhangban voltak a szakirodalom legutóbbi esetsorozatában leírtakkal: újszülöttkori megjelenés (33%), mikrocefália (66%), pszichomotoros retardáció (100%), spaszticitás (100%), súlyos értelmi fogyatékosság (66%) és a koponya CT meszesedése (66%), bár csak egy betegnek volt epilepsziás rohama.
az Aicardi-Gouti Kb szindrómában szenvedő betegek jellemzői.
| 1. ügy | 2. ügy | ügy 3 | |
|---|---|---|---|
| genetika | homozigóta mutáció (p. Ala177Thr) az RNASEH2B génben | homozigóta mutáció (341G>A) A TREX1 génben | heterozigóta mutáció (c.992C>G és p.Thr331Arg) az IFIH1 génben |
| jelenlegi életkor | 3 év | 7 év és 4 hónap | 12 év és 11 hónap |
| nem | férfi | nő | férfi |
| Származási hely | Románia | Spanyolország | Olaszország |
| AP | – | 36. hét: intrauterin növekedési korlátozás 37. hét: mikrocefália, placenta meszesedés |
szájpadhasadék |
| klinikai tünetek | |||
| életkor kezdetekor | 10 hónap | születés | 2 év |
| kezdeti bemutatás | ingerlékenység pszichomotoros regresszió |
remegés, hipotónia, gyenge sírás, növekedési kudarc | motoros készség késés |
| pszichomotoros retardáció | Igen | Igen | Igen |
| nyelv | 2-szótag words | No | Yes |
| Microcephaly | Yes | Yes | No |
| Chilblains | No | Yes | Yes |
| Epilepsy | No | Yes | No |
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFC IV |
| mozgászavar | nem | Igen | nem |
| rendellenes szemmozgások | nem | nem | nem |
| látásromlás | nem | – | Myopia |
| glaukóma | nem | nem | nem |
| halláskárosodás | – | – | nem |
| cardialis érintettség | nem | enyhe tricuspid és mitralis regurgitáció | nem |
| visszatérő láz | nem | nem | nem |
| értelmi fogyatékosság | Igen | igen, súlyos | igen, enyhe |
| Egyéb | – | – | Singleton-Merten szindróma: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
| CSF | |||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – |
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – |
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – |
| INF alfa (IU/mL) | 25 ( | 75 ( | – |
| Blood | |||
| Interferon signature | – | – | Gene overexpression |
| Neuroimaging | |||
| Calcifications on CT | No | Yes. Bazális és periventricularis ganglionokban | Igen. Szimmetrikus meszesedés a frontális régió és a lentiform nucleus mély WM-jében |
| fej MRI | diffúz és foltos változások mindkét agyfélteke WM intenzitásában, hiperintenzitás a T2 – n. Subcorticalis WM (az U rostok megkímélése) és periventricularis WM | generalizált WM érintettség a lobar WM túlsúlyával, beleértve a frontális, temporális és occipitalis lebeny subcorticalis U rostjait, kétoldalúan és szimmetrikusan, kérgi érintettség nélkül | – |
CSF, cerebrospinális folyadék; CT, számítógépes tomográfia; GMFCS, bruttó motorfunkció osztályozási rendszer; INF, interferon; IUGR, intrauterin növekedési korlátozás; MRI, mágneses rezonancia képalkotás; PNP, polyneuropathia; WM, fehér mater.
mint korábban megjegyeztük, az RNASEH2B gén homozigóta mutációi a leggyakoribb variánsok, amelyek AGS-t okoznak, fenotípusos expressziójuk általában a legjobban megfelel a klasszikus megjelenésnek.4 ez volt a helyzet a vizsgálatunkban szereplő, ilyen mutációt hordozó beteg esetében, aki 10 hónapos korában ingerlékenységgel és pszichomotoros retardációval, valamint jellegzetes neuroimaging és CSF eredményekkel jelentkezett.
az AGS eseteinek húsz százaléka újszülöttkori megjelenéssel járhat, a betegség kialakulásával a méhben.A fent említett 7 gén bármelyikének 5 mutációja vezethet ehhez a fenotípushoz, de ez a korai megjelenés leggyakrabban a TREX génhez kapcsolódik.4,5 ennek a formának a kezdeti megjelenése hasonló a TORCH fertőzéshez, hepatosplenomegáliával, hypertransaminasaemiával, thrombocytopeniával és neurológiai megnyilvánulásokkal, beleértve a szélsőséges ingerlékenységet, mozgászavarokat és epilepsziás rohamokat.5 ezeknek a betegeknek a betegsége súlyosabb, és nagyobb a halálozás kockázata. A mintánkban szereplő beteg, akinek ilyen változata volt, újszülöttkori megjelenéssel rendelkezik, jelenleg a betegség legsúlyosabb formája 3.
az ADAR1 gén, különösen az IFIH1 gén mutációi a tünetek késői megjelenésével járnak, 1 éves élet után, normális pszichomotoros fejlődéssel.5 ezen esetek némelyikében a szindróma jóindulatú, a nyelv és a motoros készségek viszonylagos megőrzésével. Az IFIH1 gén mutációjával rendelkező betegünk egyedülálló eset volt, mivel Singleton-Merten szindrómája is volt, egy ritka betegség, amelyet szintén az IFIH1 gén mutációja okozott, és amelyet fogászati diszplázia, aorta meszesedés és csontritkulás jellemez.6
célunk az AGS jelentős fenotípusos variabilitásának és a specifikus mutációkkal való összefüggésének hangsúlyozása mind a diagnózis megfontolásának ösztönzése érdekében olyan esetekben, amelyek a betegség klasszikus formájától eltérnek, mind pedig további információkkal szolgál a betegség lefolyásáról és kimenetele ezeknél a betegeknél.