periodikus Paralyses
az elsődleges periodikus paralyses klinikailag hasznos osztályozása, amelyet az 1. táblázat mutat be, magában foglalja a hypokalaemiás, hyperkalaemiás és paramyotonic formákat.
1.táblázat. Elsődleges periodikus Paralízis (jurkat-Rott és Lehmann-Horn alapján módosítva ) (táblázat megnyitása új ablakban)
|
betegség |
Gene |
fehérje |
öröklés |
mutáció |
|
HyperPP |
SCN4A |
Nav1.4 |
domináns |
finom |
|
NormoPP |
Fine (Anavar-pórus) |
|||
|
Paramyotoniacongenita |
finom |
|||
|
II típusú HypoPP |
Fine (Anavar-pórus) |
|||
|
Hypopp rendelést |
CACNA1 |
Cav1.1 |
domináns |
nyereség (6-pórus) |
|
ThyrotoxicPP |
KCNJ18 |
Kir2.18 |
domináns |
veszteség |
|
Andersen-Tawil szindróma |
KCNJ2 |
Kir2.1 |
domináns |
veszteség |
a pelyhes gyengeség fiziológiai alapja az izommembrán (azaz a sarcolemma) ingerlékenysége. A szérum káliumszintjének megváltozása nem az elsődleges PP fő hibája; a megváltozott kálium-anyagcsere a PP eredménye. Az elsődleges és tirotoxikus PP-ben petyhüdt paralízis lép fel a szérum káliumszintjének viszonylag kis változásával, míg a másodlagos PP-ben a szérum káliumszintje jelentősen abnormális.
egyetlen mechanizmus sem felelős a rendellenességek ezen csoportjáért. Így heterogének, de vannak közös vonásaik. A gyengeség általában általános, de lokalizálható. A koponyaizmok és a légzőizmok általában megkíméltek. A Stretch reflexek hiányoznak vagy csökkentek a támadások során. Az izomrostok elektromosan ingerlékenyek a támadások során. Az izomerő normális a rohamok között, de néhány év elteltével bizonyos fokú rögzített gyengeség alakul ki bizonyos típusú PP-kben (különösen az elsődleges PP-ben). Az elsődleges PP minden formája (kivéve Becker myotonia congenita) vagy autoszomális domináns öröklött vagy szórványos (valószínűleg pontmutációkból származik).
a Feszültségérzékeny ioncsatornák szorosan szabályozzák az akciós potenciál generálását (a sejtmembránok feszültségének rövid és reverzibilis változása). Ezek szelektíven és változtathatóan áteresztő ioncsatornák. Az energiafüggő ion transzporterek fenntartják a koncentrációgradienseket. Az akciós potenciálok generálása során a nátriumionok feszültségfüggő ioncsatornákon keresztül mozognak a membránon. A nyugalmi izomrost membránt elsősorban a klorid kloridcsatornákon keresztüli mozgása polarizálja, a kálium mozgása pedig repolarizálja. Nátrium, klorid és kalcium channelopathiák, mint csoport, a myotonia és a PP. A nátrium -, kalcium-és káliumcsatornák funkcionális alegységei homológ jellegűek. A nátrium-channelopathiák jobban megérthetők, mint a kalcium-vagy klorid-channelopathiák. A családi PP minden formája megmutatja a végső mechanisztikus utat, amely aberráns depolarizációt, inaktiváló nátriumcsatornákat és izomrostok ingerlékenységét foglalja magában.
ebben a cikkben elsősorban a nátrium -, kalcium-és káliumcsatornák, valamint a PP másodlagos formái foglalkoznak. A kloridcsatorna-kórok nem kapcsolódnak epizodikus gyengeséghez, és részletesebben tárgyalják a myotonikus rendellenességekről szóló cikkeket.
a csatorna diszfunkciójának összefoglalása A PP különböző típusaiban
a Hiperpp Gyors csatorna inaktiválásával a mutációk általában a transzmembrán szegmensek belső részeiben vagy az intracelluláris hurkokban helyezkednek el, amelyek befolyásolják a gyors inaktiváló részecske dokkolóhelyeit, ezáltal rontva a Gyors csatorna inaktivációt, ami tartós Na+ áramhoz vezet.
a Hipopp hiperpolarizációval aktivált kationszivárgással, amely ellensúlyozza a K + egyenirányító áramot, a mutációk a legkülső arginint vagy lizin szubsztitúciót okozzák.
Normopp depolarizációval aktivált kationszivárgás esetén a mutációk a II tartomány feszültségérzékelőjének mélyebb helyein vannak az r675 kodonnál.
az ioncsatorna diszfunkció általában jól kompenzálható normál gerjesztéssel, és gyakran további triggerek szükségesek az izmok ingerlékenységének előidézéséhez a tartós membrán depolarizáció miatt.
a glükóz – és káliumbevitel ellentétes hatást fejt ki ezekben a rendellenességekben. A HyperPP-ben a káliumbevitel kiváltja a támadást, míg a glükóz enyhíti azt. Ezzel szemben a glükóz hipokalémiás rohamokat vált ki, a kálium pedig a támadás kezelése.
vegye figyelembe az alábbi képet.
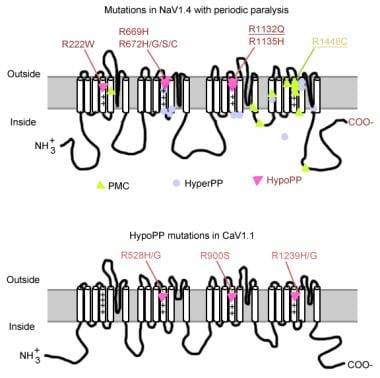 mutációk periodikus bénulásban.
mutációk periodikus bénulásban. izom nátriumcsatorna gén
a nátriumcsatorna alfa alegységgel és béta alegységgel rendelkezik. A nátriumcsatorna alfa alegysége egy 260-kd glikoprotein, amely körülbelül 1800-2000 aminosavat tartalmaz. Ez a csatorna evolúciós szempontból erősen konzervált a Drosophilától az emberig. 4 homológ doménje van (I-IV), amelyek összehajtva központi pórust képeznek, mindegyik 225-325 aminosavval rendelkezik. Minden domén 6 hidrofób szegmensből (S1-S6) áll, amelyek áthaladnak a sejtmembránon. A csatorna fő funkciói közé tartozik a feszültségérzékeny kapuzás, az inaktiválás, az ionszelektivitás. Az S5 és S6 közötti extracelluláris hurok a plazmamembránba merül, és részt vesz a pórus kialakulásában. Az S4 szegmens minden harmadik pozícióban pozitív töltésű aminosavakat tartalmaz, és feszültségérzékelőként működik. A depolarizáció során konformációs változások fordulhatnak elő, amelyek a csatorna aktiválódását és inaktiválását eredményezik. A III-S6 és a IV-S1 tartomány közötti celluláris hurok inaktiváló kapuként működik.
a nátriumcsatornának 2 kapuja van (aktiválás és inaktiválás), és 3 állapotban létezhet. A polarizált membránnal nyugalomban az aktiváló kapu zárva van, az inaktiváló kapu pedig kinyílik. A depolarizáció során az aktiválási kapu kinyílik,lehetővé téve a nátriumionok áthaladását az ioncsatornán, valamint az inaktivációs kapu dokkolóhelyét. A folyamatos depolarizációval az inaktivációs kapu bezáródik, megakadályozva a nátrium bejutását a sejtbe, és a csatorna gyors inaktivációs állapotba kerül. A csatorna ezen inaktiválása lehetővé teszi a membrán repolarizációját, ami a nyugalmi állapotba való visszatérést eredményezi az aktiváló kapu bezárásával és az inaktiváló kapu kinyitásával. Az emlősök vázizomzatában két inaktivációs folyamat fordul elő: a gyors inaktiválás magában foglalja az akciós potenciál megszüntetését, és milliszekundumos időskálán hat. A lassú inaktiválás másodpercektől percekig tart, és szabályozhatja a gerjeszthető nátriumcsatornák populációját.
a gyors és lassú inaktivációt megzavaró nátriumcsatorna-mutációk általában a Hiperpp és a myotonia fenotípusával társulnak, ahol a lassú vagy gyors inaktivációt fokozó mutációk a nátriumcsatorna-funkció elvesztését okozzák.
a nátriumcsatorna gén (SCN4A) mutációinak számos általános jellemzője van. A mutációk többsége a III és IV ismétlések közötti “inaktiváló” kapcsolóban, a IV ismétlés S4 “feszültségérzékelő” szegmensében vagy a belső membránnál található, ahol károsíthatják az inaktivációs kapu dokkolóhelyét. A klinikai fenotípus specifikus aminosav-szubsztitúcióval különbözik, és bár némi átfedés fordulhat elő a hyperkalemiás PP, A paramyotonia congenita (PC) és a kálium-súlyosbodott myotoniák (PAM) között, a 3 fenotípus általában különbözik (az alábbiakban leírtak szerint). Szinte az összes mutáns csatorna csökkentette a nátrium-áram gyors inaktiválását. A legtöbb beteg érzékeny a szisztémás káliumra vagy a hideg hőmérsékletre.
két csatorna populáció létezik, mutáns és vad típusú; a károsodott gyors inaktiváció a mutáns izomrost membránok elhúzódó depolarizációját eredményezi, és megmagyarázhatja ezeknek a rendellenességeknek a 2 kardinális tüneteit, a myotoniát és a gyengeséget. Hiperkalémiás PP-ben a mutáns csatorna kapuzásában funkciónövekedés következik be, ami megnövekedett nátriumáramot eredményez, amely túlzottan depolarizálja az érintett izomot. A myofiber membrán enyhe depolarizációja (5-10 mV), amelyet a megnövekedett extracelluláris káliumkoncentráció okozhat, azt eredményezi, hogy a mutáns csatornák nem inaktivált módban maradnak. A tartós befelé irányuló nátriumáram a vad típusú nátriumcsatornák ismétlődő tüzelését okozza, amelyet merevségnek (azaz myotonia) érzékelnek.
ha súlyosabb depolarizáció (20-30 mV) van jelen, mind a normál, mind a rendellenes csatornák inaktivációs állapotban vannak rögzítve, gyengeséget vagy bénulást okozva. Így a membrán depolarizáció súlyosságának finom különbségei különbséget tehetnek a myotonia és a bénulás között. A hőmérséklet-érzékenység a PC jellemzője. A hideg súlyosbítja a myotoniát és gyengeséget okoz. Számos mutáció kapcsolódik ehhez az állapothoz, ezek közül 3 ugyanazon a helyen (1448) az S4 szegmensben. Ezek a mutációk helyettesítik az arginint más aminosavakkal, és semlegesítik ezt az erősen konzervált S4 pozitív töltést. Ezen maradékok mutációi a PC leggyakoribb okai. A hőmérsékletérzékenységért felelős lehetséges mechanizmusok közül néhány a következő:
-
a hőmérséklet különbözőképpen befolyásolhatja a mutáns csatorna konformációs változását.
-
az alacsonyabb hőmérsékletek stabilizálhatják a mutáns csatornákat abnormális állapotban.
-
a mutációk megváltoztathatják a csatorna érzékenységét más sejtfolyamatokra, például foszforilezésre vagy második hírvivőkre.
a hiperkalémiás PP legtöbb esetben az SCN4A, a T704M és az M1592V 2 mutációjának köszönhető. A nátriumcsatorna mutációi, különösen az 1448-as és 1313-as maradékanyagoknál felelősek a paramyotonia congenita kialakulásáért. A hypokalemiás periodikus paralízis esetek kis hányada a 669 és 672 kodon (HypoPP2) mutációival függ össze. A HypoPP2-ben a nátriumcsatorna-mutációk fokozzák az inaktivációt, hogy a funkcióhiba nettó veszteségét eredményezzék.
a Normokalémiás PP mind a Hiperpp-hez (kálium érzékenység), mind a HypoPP-hez (támadások időtartama) hasonlít, és SCN4A mutációk okozzák a DII feszültségérzékelő mélyebb helyén a 675 kodonnál. Az R675 mutációk abban különböznek a HypoPP-től, hogy ezek a mutációk depolarizációval aktivált kapuzó pórusokat eredményeznek, amelyek fordított feszültségfüggőséggel generálják az áramot, mivel ez a hely erősebb depolarizációnál extracelluláris helyeknek van kitéve.
kalciumcsatorna gén
a kalciumcsatorna gén (CACNL1A3) 5 alegység (alfa-1, alfa-2, béta, gamma és delta) komplexe. A vázizom-dihidropiridin (DHP) receptor elsősorban a keresztirányú tubuláris membránban helyezkedik el. Az alfa-1 alegység kötőhelyekkel rendelkezik a DHP gyógyszerek számára, és vezeti a lassú L-típusú kalciumáramot. Részt vesz az excitation-contraction (EC) kapcsolásban is, és feszültségérzékelőként működik a szarkoplazmatikus retikulum ryanodin receptorával (azaz kalcium-felszabadító csatornával) való kapcsolatán keresztül. A membránpotenciál bármilyen változása az intracelluláris kalcium felszabadulásához kapcsolódik, lehetővé téve az EC kapcsolást. A DHP receptor/kalciumcsatorna alfa-1 alegységének pontmutációi hipokalémiás PP-t (HypoPP1) okoznak. A cacna1s gén két mutációja, az R528H és az R1239H felelős a hipokalémiás PP legtöbb esetéért.
a betegség fiziológiai alapja még mindig nem ismert, de valószínűbb, hogy a gerjesztés kudarca, nem pedig az EC kapcsolás kudarca miatt. A hipokalémia által kiváltott depolarizáció azonban csökkentheti a kalcium felszabadulását, ami közvetlenül vagy közvetve befolyásolja a csatorna feszültségszabályozását a nátriumcsatorna inaktiválásával. Az inzulin és az adrenalin hasonló módon hathat. A kalciumcsatorna gén mutációi némi hasonlóságot mutatnak az SCN4A mutációkkal. A mutációk módosítják a csatorna inaktiválását, de nem feszültségfüggő aktiválást. Az érintett betegek myotube kultúráinak felvételei 30% – kal csökkentették a DHP-érzékeny L-típusú kalciumáramot. A csatornákat alacsony membránpotenciál mellett inaktiválják.
a kalciumcsatorna-mutációk funkcióvesztést okoznak, ami csökkentett áramsűrűség és lassabb inaktiváció formájában nyilvánul meg. Nem ismert, hogy ez az inaktiváció hogyan kapcsolódik a hypokalaemia által kiváltott rohamokhoz. Legalább az R528H mutáció esetén lehetséges másodlagos csatornalopátia lép fel, amely az ATP-érzékeny káliumáram csökkentéséhez kapcsolódik a megváltozott kalcium homeosztázisból. A CACNL1A3 mutációkhoz kapcsolódó alacsonyabb áramok kissé megváltoztathatják az intracelluláris kalcium homeosztázist, ami befolyásolhatja a K+ csatornák tulajdonságait és expresszióját, különösen a KATP (ATP-érzékeny káliumcsatorna), amely a csatornák befelé egyenirányító osztályába tartozik. Az inzulin a HypoPP – ben is hat, csökkentve ezt a befelé irányuló egyenirányítót K+ áram.
a feszültségérzékelő töltésvesztesége a HypoPP legtöbb esetét okozza. A nátrium – és kalciumcsatornák homológ pórusképző alfa alegységekkel rendelkeznek. A cacnl1a3 és az SCN4A pontmutációi befolyásolják az argentin maradványokat ezen csatornák S4 feszültségérzékelőiben. Az S4 szegmensek arginin mutációi felelősek a HypoPP esetek 90% – áért.
a feszültségérzékelő töltésvesztesége a HypoPP legtöbb esetét okozza. A nátrium – és kalciumcsatornák homológ pórusképző főegységekkel rendelkeznek. Szinte az összes Cav1.1 (HypoPP-1) és Nav1.4 (HypoPP-2) mutáció semlegesíti a pozitív töltésű aminosavat a feszültségérzékelők egyik legkülső argininjében vagy lizinjében. A Nav1.4 a mutációk leggyakrabban az I, II és III ismétlések feszültségérzékelőiben helyezkednek el, ami kationszivárgást okoz.
a legkülső arginin helyettesítése egy kisebb aminosavval, például glicinnel vezetőképes utat nyit meg hiperpolarizált potenciál, ami egy befelé irányuló kationáramot eredményez (kationszivárgás vagy a (6 -)–tól az ionvezető póruson keresztül történő megkülönböztetéshez szükséges (!) a monovalens kationok hiperpolarizációval aktivált áramát jelenti S4 kapuzó pórusok ellensúlyozva az egyenirányító k+ áramokat) depolarizálja vagy destabilizálja a nyugalmi potenciált.
az S4 szegmens kifelé mozog a vezető utat lezáró depolarizáció során. A súlyos feszültségérzékelő mutációkkal rendelkező izomrostok depolarizálódnak nemcsak a hypokalaemia során, hanem a normál tartományban lévő káliumszintnél is, magyarázva az interictalis és az állandó gyengeséget. Súlyos myopathia az izomszövet zsíros pótlásával általában Cav1.1 R1239H (DIV mutációk) betegeknél fordul elő.
a glükokortikoszteroidok az inzulin és az amilin által mediált Na+ K+ ATPáz stimulálásával HypoPP-t okoznak.
káliumcsatorna gén
a befelé történő helyesbítés a Kir csatornák fontos tulajdonsága. Az egyenirányítás magában foglalja a pórusok feszültségfüggő vezetését-a pórusok blokkolását poliaminokkal és Mg++-val a depolarizáció során, és ezt az elzáródást eltávolítják a potenciális gradiens során a hiperpolarizáció során. A káliumcsatorna-mutációk az Andersen-Tawil-szindrómában és a tirotoxikus PP-ben fordulnak elő.
az Andersen-Tawil-szindrómát a diszmorfikus jellemzők, a periodikus bénulás és a szívritmuszavarok hármasa jellemzi. Ez a szindróma a KCNJ2 gén mutációival jár. A KCNJ2 gén kódolja a befelé egyenirányító káliumcsatornát Kir2.1. A kcne3 káliumcsatorna-mutációi hipokalémiás PP-t okoznak,de ezt nem igazolták.
a Kir2.6 mutációi érzékenységet okoznak a tirotoxikus PP-vel szemben. A tirotoxikus PP-ben tapasztalt epizodikus gyengeség hasonló a HypoPP és az Andersen-Tawil-szindrómában tapasztalthoz. Ez a betegség leginkább az ázsiaiak és a Latin-amerikai férfiak körében fordul elő. A tirotoxikus PP egy genetikai rendellenesség, amelyet a tirotoxikózis leplez. A Kir2.6 elsősorban a vázizomban expresszálódik. A trijód-tironin fokozza a KCNJ18 transzkripciót, ami a kir2.6 fokozott expresszióját eredményezheti. A PKC a megnövekedett PIP2 forgalom miatt aktiválódik a tirotoxikózis során, és a KIR csatornák közvetlenül kölcsönhatásba lépnek a PIP2-vel a normál kapuzás során. Andersen-Tawil-szindrómában csökkent a PIP2 affinitás. A tirotoxikus PP-ben egyik mutáció sem változtatja meg a Kir2.6 helyesbítését.