Ginecologia e ostetricia Case report
Parole chiave
Tetra-amelia; Malformazione; Genotipo; Fenotipo.
Introduzione
Le anomalie degli arti costituiscono un importante gruppo di patologie congenite caratterizzate da ipoplasia o aplasia completa di una o più ossa degli arti. Anomalie degli arti di tutti i tipi si verificano in circa 1 su 1.300 a 2.000 nascite. Queste anomalie degli arti possono essere isolate o associate ad altre anomalie . La sindrome di Tetraamelia è rara e rimangono aree grigie.
Segnaliamo due casi di tetra-amelia in una maternità di livello II a Dakar (Senegal) con sintomi simili a tetraamelia-1 (cromosoma 17q21), tetraamelia-2 (cromosoma 8q23) e sindrome di Robert (cromosoma 8p21). Questo illustra la difficoltà nel correlare fenotipo e geni coinvolti.
Segnalazioni di casi
Caso 1
La signora AD era una madre di 44 anni di cui al nostro dipartimento a 36 settimane di gestazione con grave pre-eclampsia e anomalie fetali. Aveva cinque anni, senza precedenti di anomalie fetali. Non fumava ora e non aveva mai né fumato né bevuto alcol. Non era stata esposta al fumo passivo. Era in un matrimonio consanguineo di terzo grado per tutti i suoi figli. La signora AD era risultata negativa per epatite B, HIV e sifilide. Era protetta dal virus della rosolia e non aveva precedenti esposizioni al Toxoplasma gondii. Monitoraggio ecografico eseguito in ritardo a 33 settimane e 35 settimane di gestazione recuperato oligoamniosi e idrocefalo, nonché agenesia degli arti. Le prescrizioni durante la gravidanza includevano la somministrazione di ferro e acido folico, nonché la somministrazione di sulfadoxina pirimetamina. Quest’ultimo è stato prescritto a 18 settimane e poi a 26 settimane come parte della politica di profilassi anti-malaria per le donne in gravidanza. L’altezza symphyseal-fundal misurata 28 cm. A causa di gravi caratteristiche di pre-eclampsia; è stata ricoverata immediatamente e osservata in un’unità di lavoro e consegna. Ha quindi inizialmente ricevuto IV solfato di magnesio per prevenire l’eclampsia e farmaci antipertensivi per mantenere la pressione sistolica inferiore a 160 mmHg e la pressione diastolica inferiore a 105 mmHg.
È stata presa la decisione di un parto cesareo immediato. Un grammo 2,150 nato vivo è stato estratto che successivamente è morto entro 10 minuti. Prima della morte, il corpo presentava movimenti striscianti. Sono state identificate diverse anomalie esterne (Figura 1) tra cui agenesia completa di tutti e quattro gli arti, idrocefalo con una circonferenza cranica di 39 cm. Sul viso, c’era ipertelorismo con un coloboma delle palpebre, lieve esoftalmo e aniridia. La bocca era simile a una V rovesciata senza una chiara delimitazione delle labbra e il naso era rudimentale. Il neonato era privo di tegumenti (capelli e sopracciglia). Le orecchie erano ridotte a schizzi che sembravano fessure. Il collo era corto. Il tronco era ridotto a una struttura conica lunga 26 cm con un cordone ombelicale all’estremità inferiore. Il petto era stretto. Appena sotto l’ombelico, il tronco allargato all’indietro era presente al pavimento del polo caudale, una linea mediana con recessioni e un germogliamento che può corrispondere ad un fallo di tipo indeterminato. Sono state notate agenesia del bacino, genitali e imperforazione anale. La patologia fetale non è stata eseguita. Tuttavia, la morte entro 10 minuti dal parto e l’aspetto conico del torace possono suggerire anomalie polmonari.
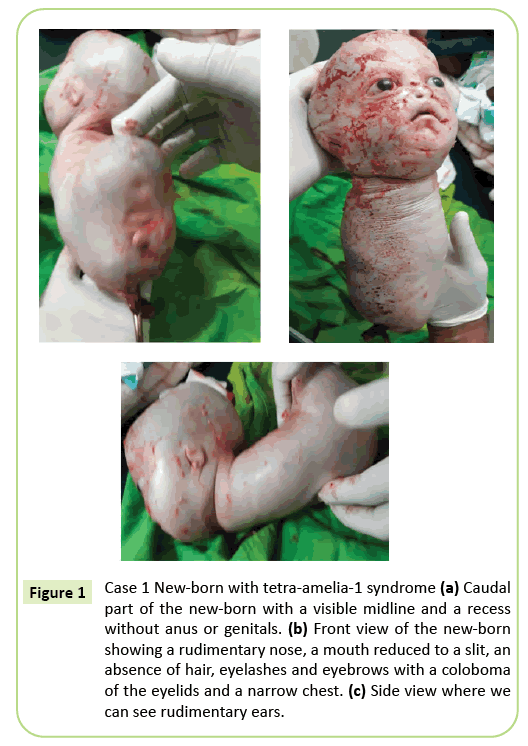
Figura 1: Caso 1 Neonato con sindrome di tetra-amelia-1 (a) Parte caudale del neonato con una linea mediana visibile e una rientranza senza ano o genitali. (b) Vista frontale del neonato che mostra un naso rudimentale, una bocca ridotta a una fessura, un’assenza di capelli, ciglia e sopracciglia con un coloboma delle palpebre e un petto stretto. (c) Vista laterale dove possiamo vedere orecchie rudimentali.
Caso 2
Il secondo caso è stato un 22-year-old primigravida riferito alla nostra struttura per l’ecografia a 37 settimane di gestazione. Non era in un matrimonio consanguineo. Era risultata negativa all’epatite B, all’HIV e alla sifilide. Non è stata testata per la toxoplasmosi e la rosolia. Nessun monitoraggio ecografico è stato fatto durante la gravidanza. L’esame clinico è stato coerente con ritardo della crescita fetale (altezza del fondo: 26 cm). I risultati degli ultrasuoni hanno mostrato che l’omero era distorto misurando 23,9 mm corrispondenti a 17 settimane di gestazione. C’era agenesia del femore. Le ali iliache erano visibili sugli ultrasuoni. Non sono state identificate anomalie polmonari o cardiache. La consegna è stata avviata. Il neonato aveva un fenotipo femminile con un punteggio Apgar di 9 al 5 ° minuto. La morfologia della testa e del tronco era senza particolarità. Gli arti superiori erano ridotti a due ceppi lunghi 3 cm. È stata notata agenesia completa dei 2 arti inferiori. Era un’anomalia simmetrica (Figura 2).
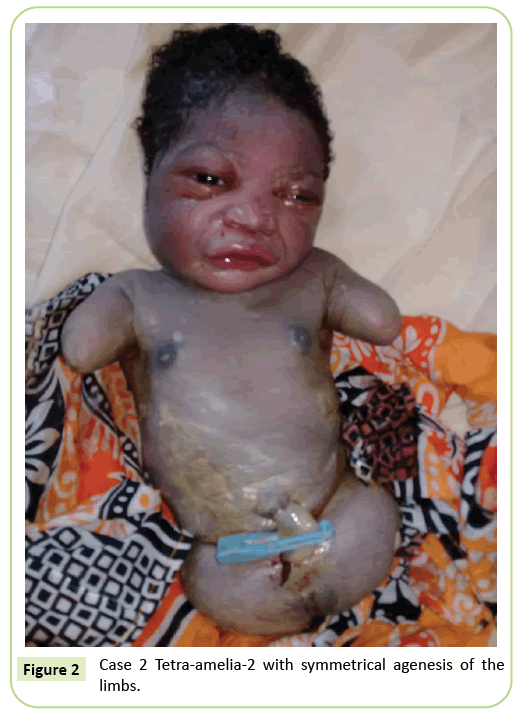
Figura 2: Caso 2 Tetra-amelia-2 con agenesia simmetrica degli arti.
Discussione
Bermejo-Sanchez et al. descritta nel 2011 l’epidemiologia di amelia congenita utilizzando i dati raccolti da 20 programmi di sorveglianza delle anomalie congenite, provenienti da tutti i continenti tranne l’Africa, tra il 1968 e il 2006. In totale, sono stati identificati 326 casi di amelia tra 23.110.591 nati vivi, nati morti e aborti. La prevalenza è stata di 1,41/100.000 .
Tetra-amelia si riferisce alla completa assenza degli arti e si verifica più raramente. A nostra conoscenza, tetra-amelia-1 è descritto in 7 famiglie. Sembra seguire un’eredità autosomica recessiva. In tutte le famiglie, tetra-amelia-1 è stato associato a gravi malformazioni delle altre parti del corpo tra cui viso e testa, anomalie del sistema nervoso, scheletro e genitali. I polmoni erano sottosviluppati in molti casi, il che rende la respirazione difficile o impossibile . Zimmer et al. riportato nel 1985 una famiglia fortemente inbred in cui 6 neonati avevano tetra-amelia-1 e idrocefalo. Hanno descritto in uno dei feti una totale assenza di osso pelvico, labbro leporino e palato, arrhinia e aplasia delle orecchie. Sono stati anche notati un polmone sinistro bilaterale, un canale arterioso persistente, un’imperforazione anale. I test fetali hanno eliminato la diagnosi della sindrome di Robert . Altri casi recuperati in letteratura includono quello di Kosaki et al., nel 1996, con un feto di cariotipo 46, XX con tetrafocomelia e grave ipoplasia polmonare oltre a anomalie del viso e della testa . Rosenak et al. descritto un caso di tetra-amelia con grave ipoplasia polmonare in due feti di una coppia non consanguinea. I test fetali hanno escluso la diagnosi della sindrome di Robert . Altri due casi sono stati segnalati da Zlotogora et al. nel 1993. Entrambi i pazienti sono morti subito dopo la nascita e gli autori hanno suggerito l’esistenza di ipoplasia polmonare. Niemann et al. segnalato una famiglia turca consanguinea in cui 4 degli 8 fratelli soffrivano di tetra-amelia. Oltre all’assenza dei 4 arti, gli esami fetali di 3 feti hanno rivelato anomalie multiple: labbra schisi e /o palatine, laparoschisi, anomalie polmonari, ipoplasia del bacino, atresia delle choane, imperforazione vaginale e anale . Infine, nel 2005, Krahn et al. descritto 2 fratelli nati da genitori consanguinei affetti da tetraamelia e grave ipoplasia polmonare. Le clavicole e le scapole erano normali nel secondo feto. Il cariotipo era normale .
La sindrome di Tetra-amelia-1 o TETAMS1 è causata da una mutazione omozigote nel gene WNT3 sul cromosoma 17q21 con ereditarietà autosomica recessiva. La sindrome di Tetraamelia-2 (TETAMS2) è caratterizzata da arti rudimentali o da una completa assenza degli arti, generalmente simmetrici e in alcuni casi agenesia bilaterale dei polmoni. Sono anche usuali anomalie del sistema vascolare polmonare e dismorfie tra cui labbro leporino bilaterale e palato, anchiloglossia, ipoplasia mandibolare, microretrognazia e aplasia labioscrotale .
Szenker-Ravi, studiando 4 famiglie di tetra-amelia con agenesia o ipoplasia polmonare, ha notato un’eterogeneità fenotipica con anomalie degli arti di varia gravità . Il sequenziamento degli esomi in queste 4 famiglie ha permesso di identificare mutazioni omozigoti troncanti nel gene RSPO2 . La sindrome di Tetraamelia-2 è causata da una mutazione omozigote nel gene RSPO2 (610575) situato sul cromosoma 8q23 .
Il fenotipo del primo caso descritto in questo articolo corrisponde ad una sindrome di tetra-amelia-1 dovuta in particolare alla presenza di idrocefalo, anomalie genitali e un naso rudimentale. Il torace stretto e la morte precoce prima del decimo minuto di vita suggeriscono una grave ipoplasia polmonare. Questo caso evidenzia l’eterogeneità fenotipica con un coloboma palpebrale, ipertelorismo, esoftalmo e appendici rare.
Consideriamo il secondo caso nel nostro studio una sindrome tetramelia-2 considerando la tetra-amelia simmetrica con presenza di ceppi degli arti superiori. La diagnosi di tetra-amelia deve essere effettuata precocemente durante il monitoraggio ecografico. Pertanto, la consapevolezza dovrebbe essere sollevata circa l’importanza del monitoraggio ecografico e l’uso di 3D/4D per migliorare i risultati di screening. La diagnosi di una massa pelvica su ultrasuoni in coppia con amelia dovrebbe sollevare sospetti per la sindrome da difetto degli arti di fusione spleno-gonadica.
Inoltre, l’esame fetale e il test fetale utilizzando le tecnologie in evoluzione del microarray cromosomico e del sequenziamento dell’esoma e del genoma devono essere incoraggiati nelle nostre impostazioni. Una migliore caratterizzazione dei casi consente di fornire consulenza alle coppie e una migliore conoscenza di queste anomalie cliniche.
Conclusione
La sindrome di Tetra-amelia è scarsa e rimangono ancora aree grigie. Questi due casi, rispetto a quanto già descritto in letteratura, illustrano l’eterogeneità fenotipica della tetraamelia. Data la rara incidenza di queste anomalie, sarebbe importante creare un registro internazionale delle anomalie al fine di segnalare i casi e istituire una banca di campioni per gli studi genetici estesi ai genitori.
- Wilcox WR, Coulter CP, Schmitz ML (2015) Disturbi congeniti da carenza di arti. Clin Perinatol 42: 281-300.
- Bermejo SE, Cuevas L, Amar E (2011) Amelia: uno studio epidemiologico descrittivo multicentrico in un ampio set di dati dell’International Clearinghouse for Birth Defects Surveillance and Research e panoramica della letteratura. Am J Med Genet C Semin Med Genet 157: 288-304.
- Zlotogora JSM, Shabany YO, Jarallah RY (1993) Sindrome di tetraamelia con ipoplasia polmonare. Am J Med Genet 47: 570-571.
- Zimmer EZ (1985) Tetra-amelia con malformazioni multiple in sei feti maschi in un unico parente. Europ. J Pediat 144: 412-414.
- Rosenak D (1991) Tetraamelia ricorrente e ipoplasia polmonare con malformazioni multiple nelle sib. Am J Med Genet 38: 25-28.
- Gershoni BR (1990) Sindrome di Roberts o “Amelia X-linked” ? . Am J Med Genet 37: 569-572.
- Kosaki K, Jones MC, Stayboldt C (1996) Focomelia di Zimmer: delineazione mediante analisi delle coordinate principali. Am J Med Genet 66: 55-59.
- La mutazione omozigote WNT3 di Niemann S(2004) causa la tetra-amelia in una grande famiglia consanguinea. Am J Hum Genet 74: 558-563.
- Krahn M (2005)Sindrome di Tetra-amelia e aplasia polmonare: relazione di una nuova famiglia ed esclusione di geni candidati. Clin Genet 68: 558-560.
- Szenker-RE, Altunoglu U (2018) RSPO2 inibizione di RNF43 e ZNRF3 governa lo sviluppo degli arti indipendentemente da LGR4/5/6. Natura 557: 564-569.