Paralisi periodiche
Una classificazione clinicamente utile delle paralisi periodiche primarie, mostrata nella Tabella 1, include forme ipokalemiche, iperkalemiche e paramyotoniche.
Tabella 1. Primaria Paralisi Periodica (modificato da Jurkat-Rott e Lehmann-Corno ) (Tabella Aperta in una nuova finestra)
|
Malattia |
Gene |
Proteina |
Eredità |
Mutazione |
|
HyperPP |
SCN4A |
Nav1.4 |
Dominante |
Bene |
|
NormoPP |
Fine (ω-poro) |
|||
|
Paramyotoniacongenita |
Bene |
|||
|
HypoPP di Tipo II |
Fine (ω-poro) |
|||
|
HypoPP PER |
CACNA1S |
Cav1.1 |
Dominante |
Guadagno (ω-poro) |
|
ThyrotoxicPP |
KCNJ18 |
Kir2.18 |
Dominante |
Perdita |
|
Andersen-Tawil, sindrome di |
KCNJ2 |
Kir2.1 |
Dominante |
Perdita |
La base fisiologica di debolezza flaccido è inexcitability della membrana muscolare (vale a dire, il sarcolemma). L’alterazione del livello di potassio sierico non è il difetto principale nel PP primario; il metabolismo alterato del potassio è il risultato del PP. Nel PP primario e tireotossico, la paralisi flaccida si verifica con cambiamenti relativamente piccoli nel livello di potassio sierico, mentre nel PP secondario i livelli di potassio sierico sono marcatamente anormali.
Nessun singolo meccanismo è responsabile di questo gruppo di disturbi. Pertanto, sono eterogenei ma condividono alcuni tratti comuni. La debolezza di solito è generalizzata ma può essere localizzata. La muscolatura cranica e i muscoli respiratori di solito sono risparmiati. I riflessi di stiramento sono assenti o diminuiti durante gli attacchi. Le fibre muscolari sono elettricamente ineccitabili durante gli attacchi. La forza muscolare è normale tra gli attacchi ma, dopo alcuni anni, si sviluppa un certo grado di debolezza fissa in alcuni tipi di PP (specialmente PP primario). Tutte le forme di PP primario (eccetto myotonia congenita di Becker) sono ereditate autosomiche dominanti o sporadiche (molto probabilmente derivanti da mutazioni puntiformi).
I canali ionici sensibili alla tensione regolano strettamente la generazione di potenziali d’azione (alterazioni brevi e reversibili della tensione delle membrane cellulari). Questi sono canali ionici selettivamente e variabilmente permeabili. I trasportatori di ioni dipendenti dall’energia mantengono gradienti di concentrazione. Durante la generazione di potenziali d’azione, gli ioni sodio si muovono attraverso la membrana attraverso canali ionici voltaggio-dipendenti. La membrana di fibra muscolare a riposo è polarizzata principalmente dal movimento del cloruro attraverso i canali del cloruro ed è ripolarizzata dal movimento del potassio. Le canalopatie di sodio, cloruro e calcio, come gruppo, sono associate a miotonia e PP. Le subunità funzionali dei canali del sodio, del calcio e del potassio sono omologhe. Le canalopatie del sodio sono meglio comprese delle canalopatie del calcio o del cloruro. Tutte le forme di PP familiare mostrano il percorso meccanicistico finale che coinvolge la depolarizzazione aberrante, i canali del sodio inattivanti e l’ineccitabilità delle fibre muscolari.
La discussione in questo articolo riguarda principalmente le canalopatie di sodio, calcio e potassio, nonché le forme secondarie di PP. Le canalopatie del cloruro non sono associate a debolezza episodica e sono discusse in modo più dettagliato negli articoli sui disturbi miotonici.
Riassunto della disfunzione del canale in vari tipi di PP
Con l’inattivazione del canale veloce HyperPP, le mutazioni si trovano solitamente nelle parti interne dei segmenti transmembrana o nei loop intracellulari che interessano i siti di attracco per la particella inattivante veloce, compromettendo così l’inattivazione del canale veloce che porta alla corrente Na+ persistente.
Con ipopp iperpolarizzazione attivato perdita di catione contrastare K+-rettifica corrente, mutazioni causano arginina o lisina sostituzione più esterna.
Con NormoPP depolarizzazione attivato catione perdita, mutazioni sono in posizioni più profonde del sensore di tensione del dominio II a codone R675.
La disfunzione del canale ionico è solitamente ben compensata con l’eccitazione normale e spesso sono necessari ulteriori trigger per produrre ineccitabilità muscolare a causa della depolarizzazione prolungata della membrana.
L’assunzione di glucosio e potassio ha gli effetti opposti in questi disturbi. In HyperPP, l’assunzione di potassio innesca l’attacco, mentre il glucosio lo migliora. Al contrario, il glucosio provoca attacchi ipokalemici e il potassio è il trattamento per l’attacco.
Nota l’immagine qui sotto.
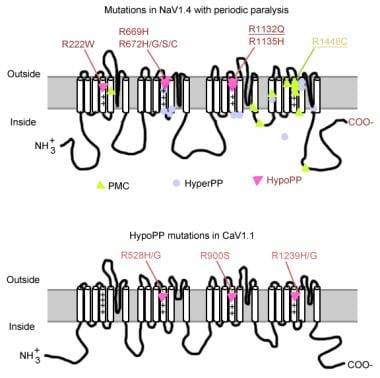 Mutazioni nella paralisi periodica.
Mutazioni nella paralisi periodica. Gene del canale del sodio muscolare
Il canale del sodio ha una subunità alfa e una subunità beta. La subunità alfa del canale del sodio è una glicoproteina 260-kd comprendente circa 1800-2000 aminoacidi. Questo canale è altamente conservato evolutivamente dalla Drosophila all’uomo. Ha 4 domini omologhi (I-IV) che si piegano per formare un poro centrale, ciascuno con 225-325 aminoacidi. Ogni dominio è costituito da 6 segmenti idrofobici (S1-S6) che attraversano la membrana cellulare. Le funzioni principali del canale includono gating sensibile alla tensione, inattivazione e selettività ionica. Il ciclo extracellulare tra S5 e S6 si immerge nella membrana plasmatica e partecipa alla formazione del poro. Il segmento S4 contiene aminoacidi caricati positivamente in ogni terza posizione e funziona come un sensore di tensione. Cambiamenti di conformazione possono verificarsi durante la depolarizzazione, con conseguente attivazione e inattivazione del canale. Il ciclo cellulare tra dominio III – S6 e dominio IV-S1 agisce come un cancello inattivante.
Il canale del sodio ha 2 porte (attivazione e inattivazione) e può esistere in 3 stati. A riposo con la membrana polarizzata, il gate di attivazione viene chiuso e il gate di inattivazione viene aperto. Con la depolarizzazione, il cancello di attivazione si apre, consentendo agli ioni di sodio di passare attraverso il canale ionico e anche di esporre un sito di attracco per il cancello di inattivazione. Con la continua depolarizzazione, il cancello di inattivazione si chiude, bloccando l’ingresso di sodio nella cellula e facendo entrare il canale nello stato di inattivazione rapida. Questa inattivazione del canale consente alla membrana di essere ripolarizzata, con conseguente ritorno allo stato di riposo con il cancello di attivazione chiuso e il cancello di inattivazione aperto. Due processi di inattivazione si verificano nel muscolo scheletrico dei mammiferi: l’inattivazione rapida comporta la cessazione del potenziale d’azione e agisce su una scala temporale di millisecondi. L’inattivazione lenta richiede secondi o minuti e può regolare la popolazione dei canali del sodio eccitabili.
Le mutazioni del canale del sodio che interrompono l’inattivazione rapida e lenta sono solitamente associate a un fenotipo di HyperPP e miotonia, dove le mutazioni che aumentano l’inattivazione lenta o veloce producendo la perdita della funzione del canale del sodio causano ipopp.
Le mutazioni del gene del canale del sodio (SCN4A) hanno diverse caratteristiche generali. La maggior parte delle mutazioni si trovano nel linker “inattivante” tra le ripetizioni III e IV, nel segmento “voltage-sensing” S4 della ripetizione IV o nella membrana interna dove potrebbero compromettere il sito di docking per il gate di inattivazione. Il fenotipo clinico differisce dalla sostituzione specifica dell’amminoacido e, mentre una certa sovrapposizione può accadere fra il PP hyperkalemic, paramyotonia congenita (PC) e myotonias potassio-aggravato (PAM), i 3 fenotipi sono generalmente distinti (come descritto sotto). Quasi tutti i canali mutanti hanno alterato l’inattivazione rapida della corrente di sodio. La maggior parte dei pazienti è sensibile al potassio sistemico o alla temperatura fredda.
Esistono due popolazioni di canali, mutanti e wild-type; l’alterata inattivazione rapida provoca una prolungata depolarizzazione delle membrane delle fibre muscolari mutanti e può spiegare i 2 sintomi cardinali di questi disturbi, miotonia e debolezza. Nel PP iperkalemico, un guadagno di funzione si verifica nel gating del canale mutante, con conseguente aumento della corrente di sodio che depolarizza eccessivamente il muscolo interessato. La depolarizzazione delicata (5-10 mV) della membrana del miofibero, che può essere causata dalle concentrazioni extracellulari aumentate del potassio, provoca i canali mutanti che sono mantenuti nel modo non inactivated. La persistente corrente di sodio verso l’interno causa la cottura ripetitiva dei canali del sodio wild-type, che viene percepita come rigidità (cioè miotonia).
Se è presente una depolarizzazione più grave (20-30 mV), sia i canali normali che quelli anormali sono fissati in uno stato di inattivazione, causando debolezza o paralisi. Pertanto, sottili differenze nella gravità della depolarizzazione della membrana possono fare la differenza tra miotonia e paralisi. La sensibilità alla temperatura è un segno distintivo del PC. Il freddo esacerba la miotonia e induce debolezza. Un certo numero di mutazioni sono associate a questa condizione, 3 delle quali nello stesso sito (1448) nel segmento S4. Queste mutazioni sostituiscono l’arginina con altri amminoacidi e neutralizzano questa carica positiva S4 altamente conservata. Le mutazioni di questi residui sono la causa più comune di PC. Alcuni dei possibili meccanismi responsabili della sensibilità alla temperatura includono quanto segue:
-
La temperatura può influenzare in modo differenziato il cambiamento conformazionale nel canale mutante.
-
Temperature più basse possono stabilizzare i canali mutanti in uno stato anormale.
-
Le mutazioni possono alterare la sensibilità del canale ad altri processi cellulari, quali la fosforilazione o i secondi messaggeri.
La maggior parte dei casi di PP iperkalemico sono dovuti a 2 mutazioni in SCN4A, T704M e M1592V. Le mutazioni nel canale del sodio, specialmente ai residui 1448 e 1313, sono responsabili della paramyotonia congenita. Una piccola percentuale di casi di paralisi periodica ipokalemica è associata a mutazioni ai codoni 669 e 672 (ipopp2). In HypoPP2, le mutazioni del canale del sodio migliorano l’inattivazione per produrre una perdita netta di difetto di funzione.
Normokalemic PP assomiglia sia HyperPP (potassium sensitivity) che HypoPP (duration of attacks) ed è causato da mutazioni SCN4A in una posizione più profonda del sensore di tensione DII al codone 675. Le mutazioni R675 differiscono da HypoPP in quanto queste mutazioni provocano la depolarizzazione-attivato gating pore generando ω-corrente con dipendenza di tensione invertita come questo sito è esposto a siti extracellulari a depolarizzazione più forte.
Gene dei canali del calcio
Il gene dei canali del calcio (CACNL1A3) è un complesso di 5 subunità (alfa-1, alfa-2, beta, gamma e delta). Il recettore diidropiridina (DHP) del muscolo scheletrico si trova principalmente nella membrana tubolare trasversale. La subunità alfa-1 ha siti di legame per i farmaci DHP e conduce la lenta corrente di calcio di tipo L. Partecipa anche all’accoppiamento eccitazione-contrazione (EC) e agisce come un sensore di tensione attraverso il suo legame con il recettore ryanodine del reticolo sarcoplasmatico (cioè, canale di rilascio del calcio). Qualsiasi cambiamento nel potenziale di membrana è legato al rilascio intracellulare di calcio, consentendo l’accoppiamento EC. Mutazioni puntiformi nella subunità alfa-1 del recettore DHP/canale del calcio causano ipopotassiemia PP (HypoPP1). Due mutazioni del gene CACNA1S, R528H e R1239H, sono responsabili della maggior parte dei casi di PP ipokalemico.
La base fisiologica della malattia non è ancora compresa, ma è più probabile a causa di un fallimento dell’eccitazione piuttosto che di un fallimento dell’accoppiamento CE. Tuttavia, la depolarizzazione indotta da ipopotassiemia può ridurre il rilascio di calcio, influenzando il controllo della tensione del canale direttamente o indirettamente attraverso l’inattivazione del canale del sodio. L ‘insulina e l’ adrenalina possono agire in modo simile. Le mutazioni del gene dei canali del calcio hanno alcune somiglianze con le mutazioni SCN4A. Le mutazioni modificano l’inattivazione del canale ma non l’attivazione dipendente dalla tensione. Le registrazioni da colture di miotubi di pazienti affetti hanno rivelato una riduzione del 30% della corrente di calcio di tipo L sensibile al DHP. I canali sono inattivati a bassi potenziali di membrana.
Le mutazioni dei canali del calcio causano una perdita di funzione manifestata come una ridotta densità di corrente e un’inattivazione più lenta. Come questa inattivazione sia correlata agli attacchi indotti da ipopotassiemia non è chiaro. Almeno nella mutazione R528H, si verifica una possibile channelopathy secondaria, legata ad una riduzione della corrente di potassio ATP-sensibile dall’omeostasi alterata del calcio. Le correnti più basse associate alle mutazioni di CACNL1A3 potrebbero alterare leggermente l’omeostasi intracellulare del calcio, il che potrebbe influenzare le proprietà e l’espressione dei canali K+, in particolare KATP (ATP-sensitive potassium channel) appartenente alla classe dei canali raddrizzatori verso l’interno. L’insulina agisce anche in HypoPP riducendo questo raddrizzatore verso l’interno K+ corrente.
La perdita di carica del sensore di tensione rappresenta la maggior parte dei casi di HypoPP. I canali del sodio e del calcio hanno subunità alfa omologhe che formano pori. Mutazioni puntiformi in CACNL1A3 e SCN4A influenzano i residui argentini nei sensori di tensione S4 di questi canali. Le mutazioni di arginina nei segmenti S4 sono responsabili del 90% dei casi di IPOPP.
La perdita di carica del sensore di tensione rappresenta la maggior parte dei casi di HypoPP. I canali del sodio e del calcio hanno subunità α omologhe che formano pori. Quasi tutte le mutazioni in Cav1.1 (HypoPP-1) e Nav1.4 (HypoPP-2) neutralizzano un amminoacido caricato positivamente in una delle arginine o lisine più esterne dei sensori di tensione. Il Nav1.4 mutazioni si trovano più comunemente nei sensori di tensione delle ripetizioni I, II e III, causando una perdita di cationi.
La sostituzione dell’arginina più esterna con un amminoacido più piccolo come la glicina apre una via conduttiva a potenziale iperpolarizzato, con conseguente corrente cationica verso l’interno (perdita di cationi o corrente ω per distinguere da (ω-) attraverso il poro conduttore di ioni, è una corrente attivata dall’iperpolarizzazione di cationi monovalenti attraverso il poro di gating S4 che
Il segmento S4 si muove verso l’esterno durante la depolarizzazione chiudendo il percorso conduttivo. Le fibre muscolari con gravi mutazioni del sensore di tensione vengono depolarizzate non solo durante l’ipopotassiemia ma anche a livelli di potassio nel range normale, spiegando la debolezza interictale e permanente. Miopatia grave con sostituzione grassa del tessuto muscolare si trova comunemente nei pazienti con Cav1.1 R1239H (mutazioni DIV).
I glucocorticosteroidi causano ipopp stimolando Na + K + ATPasi mediata da insulina e amilina.
Gene del canale del potassio
La rettifica verso l’interno è una proprietà importante dei canali Kir. La rettifica comporta il blocco dei pori di conduzione dipendente dalla tensione del poro con poliammine e Mg++ durante la depolarizzazione e questo blocco viene rimosso durante il gradiente potenziale durante l’iperpolarizzazione. Mutazioni del canale del potassio sono osservate nella sindrome di Andersen-Tawil e nella PP tireotossica.
La triade delle caratteristiche dismorfiche, della paralisi periodica e delle aritmie cardiache caratterizza la sindrome di Andersen-Tawil. Questa sindrome è associata a mutazioni nel gene KCNJ2. Il gene KCNJ2 codifica il canale del potassio Kir2.1 rettificante verso l’interno. Mutazioni del canale del potassio in KCNE3 sono riportate per causare PP ipokalemico, ma questo non è stato dimostrato.
Mutazioni in Kir2. 6 causano suscettibilità alla PP tireotossica. La debolezza episodica osservata nella PP tireotossica è simile a quella osservata nella sindrome di HypoPP e Andersen-Tawil. Questo disturbo è più diffuso in asiatici e uomini latino-americani. La PP tireotossica è una malattia genetica smascherata dalla tireotossicosi. Kir2.6 è espresso principalmente nel muscolo scheletrico. La triiodotironina migliora la trascrizione KCNJ18, che può guidare l’espressione migliorata di Kir2. 6. Il PKC viene attivato durante la tireotossicosi a causa dell’aumento del turnover PIP2 e i canali Kir interagiscono direttamente con PIP2 durante il normale gating. Nella sindrome di Andersen-Tawil, vi è una diminuzione dell’affinità PIP2. In PP tireotossico, nessuna delle mutazioni altera la rettifica di Kir2. 6.