Sindrome di Aicardi-Goutières: Spettro fenotipico e genetico in una serie di tre casi / Anales de Pediatría
La sindrome di Aicardi-Goutières (AGS) è una malattia ereditaria rara la cui esatta prevalenza è sconosciuta. È stata descritta per la prima volta nel 1984 da Jean Aicardi e Francoise Goutières come un’encefalopatia progressiva con esordio nei primi mesi di vita caratterizzata da linfocitosi del liquido cerebrospinale (CSF) e calcificazioni nei gangli della base.1 Si manifesta con irritabilità, ritardo psicomotorio, spasticità, distonia, crisi epilettiche, episodi ricorrenti di febbre asettica e microcefalia. La mortalità è più elevata durante la fase encefalopatica e, sebbene la malattia si stabilizzi in genere in seguito, causa gravi sequele neurologiche. Altre caratteristiche che possono comparire durante il suo decorso sono geloni, sintomi oculari (principalmente glaucoma), coinvolgimento cardiaco o disturbi autoimmuni.2 Interferoni di tipo I svolgono un ruolo cruciale nella patogenesi di AGS, in cui la loro espressione è sovraregolata portando ad un aumento della produzione.3 Per questo motivo, uno dei classici risultati di laboratorio in questi pazienti è un livello elevato di interferone alfa nel liquido cerebrospinale, insieme a pleocitosi e livelli ugualmente elevati di neopterina e biopterina. La potenziale utilità di valutare il livello di espressione dei geni stimolati da interferone da interferone nel sangue periferico come marcatore è attualmente in fase di studio, in quanto vi è evidenza che questi livelli rimangono elevati oltre la fase encefalopatica (“firma interferone”).3-5 Un’altra caratteristica fondamentale è la rilevazione di anomalie neuroimaging tra cui calcificazioni nei gangli della base e cambiamenti nella sostanza bianca (Fig. 1). Ad oggi, sappiamo di 7 geni le cui mutazioni possono portare alla sovraregolazione della via dell’interferone: ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1 e IFIH1. Mutazioni eterozigoti sono state descritte per i geni TREX1, ADAR e IFIH1, mentre le mutazioni riportate in tutti gli altri geni sono state omozigoti.2 Mutazioni nel gene IFIH1 sono state rilevate più recentemente (2014)4 e sono quindi le varianti patogene meno frequenti, mentre le mutazioni nei geni RNASEH2B e TREX1 rappresentano la più alta percentuale di casi diagnosticati di AGS.
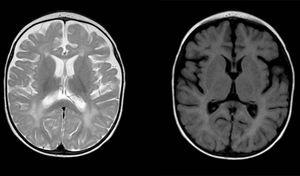
Anomalie del segnale diffuse e irregolari nella sostanza bianca in entrambi gli emisferi cerebrali, iperintenso nelle immagini ponderate in T2. Spazio subaracnoideo allargato con predominanza frontotemporale in entrambi gli emisferi, con allargamento della fessura interemisferica e aumento delle dimensioni ventricolari (in assenza di aumento della pressione), compatibile con atrofia corticale e sottocorticale.
Negli ultimi decenni, grazie ai progressi della genetica che hanno permesso la rilevazione di queste mutazioni specifiche, è emersa l’evidenza di un ampio spettro fenotipico oltre la classica presentazione basata sul gene causale. Presentiamo i casi di 3 pazienti che hanno ricevuto una diagnosi di AGS negli ultimi 8 anni con l’obiettivo di analizzare le loro caratteristiche cliniche in relazione al difetto genetico sottostante (Tabella 1). In generale, le caratteristiche di presentazione dell’AGS erano coerenti con quelle descritte nelle serie di casi più recenti in letteratura: presentazione neonatale (33%), microcefalia (66%), ritardo psicomotorio (100%), spasticità (100%), grave disabilità intellettiva (66%) e calcificazioni sulla TC cranica (66%), sebbene solo un paziente avesse crisi epilettiche.
Caratteristiche dei pazienti con sindrome di Aicardi-Goutières.
| Caso 1 | Caso 2 | Caso 3 | |
|---|---|---|---|
| Genetica | mutazione Omozigote (p.Ala177Thr) in RNASEH2B gene | mutazione Omozigote (341G>A) in TREX1 gene | mutazione Eterozigote (c.992C>G e p.Thr331Arg) in IFIH1 gene |
| età | 3 anni | 7 anni e 4 mesi | 12 anni e 11 mesi |
| Sesso | Maschio | Femmina | Maschio |
| Origine | Romania | Spagna | Italia |
| AP | – | Settimana 36: restrizione della crescita intrauterina Settimana 37: microcefalia, placentare calcificazione |
palatoschisi |
| manifestazioni Cliniche | |||
| l’Età di esordio | 10 mesi | Nascita | 2 anni |
| Presentazione iniziale | Irritabilità regressione Psicomotoria |
Tremore, ipotonia, debolezza del pianto, la mancata crescita | Motore abilità di ritardo |
| ritardo Psicomotorio | Sì | Sì | Sì |
| Lingua | 2-Sillaba words | No | Yes |
| Microcephaly | Yes | Yes | No |
| Chilblains | No | Yes | Yes |
| Epilepsy | No | Yes | No |
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFCS IV |
| disturbo del Movimento | No | Sì | No |
| Anormali movimenti oculari | No | No | No |
| Visual impairment | No | – | la Miopia |
| il Glaucoma | No | No | No |
| la perdita dell’Udito | – | – | No |
| Coinvolgimento cardiaco | No | Lieve rigurgito della tricuspide e mitrale | No |
| febbre Ricorrente | No | No | No |
| disabilità intellettiva | Sì | Sì, grave | Sì, mite |
| Altri | – | – | Singleton-Merten, sindrome di: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
| CSF | |||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – |
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – |
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – |
| INF alfa (IU/mL) | 25 ( | 75 ( | – |
| Blood | |||
| Interferon signature | – | – | Gene overexpression |
| Neuroimaging | |||
| Calcifications on CT | No | Yes. Nei gangli basali e periventricolari | Sì. Calcificazioni simmetriche in WM profondo della regione frontale e del nucleo lentiforme |
| MRI testa | Cambiamenti diffusi e irregolari nell’intensità WM di entrambi gli emisferi cerebrali, iperintenso su T2. Il coinvolgimento di sottocorticali WM (risparmiando così i U fibre) e periventricolare WM | Generalizzata WM coinvolgimento con predominanza di lobare WM tra cui il sottocorticali U fibre di frontale, temporale e occipitale lobi, bilateralmente e simmetricamente, senza interessamento corticale | – |
LIQUOR, liquido cerebrospinale; tac, tomografia; GMFCS, Gross Motor Function Sistema di Classificazione; INF, interferone; IUGR, restrizione della crescita intrauterina; risonanza magnetica, risonanza magnetica, PNP, polineuropatia; WM, bianco mater.
Come notato prima, le mutazioni omozigoti nel gene RNASEH2B sono le varianti più frequenti che causano AGS e la loro espressione fenotipica di solito si conforma di più alla presentazione classica.4 Questo è stato il caso del paziente nel nostro studio che ha portato una tale mutazione, che ha avuto inizio all’età di 10 mesi con irritabilità e ritardo psicomotorio e con risultati caratteristici di neuroimaging e CSF.
Il venti percento dei casi di AGS può avere una presentazione neonatale, con l’insorgenza della malattia che si verifica in utero.5 Mutazioni in uno qualsiasi dei 7 geni di cui sopra possono portare a questo fenotipo, ma questa presentazione precoce è più frequentemente associata al gene TREX.4,5 La presentazione iniziale di questa forma è simile a quella di un’infezione TORCH, con epatosplenomegalia, ipertransaminasemia, trombocitopenia e manifestazioni neurologiche tra cui irritabilità estrema, disturbi del movimento e crisi epilettiche.5 Questi pazienti hanno un decorso della malattia più grave e sono a più alto rischio di morte. Il paziente nel nostro campione che ha presentato una tale variante ha avuto una presentazione neonatale e attualmente ha la forma più grave di malattia dell ‘ 3.
Le mutazioni nel gene ADAR1 e in particolare nel gene IFIH1 sono associate ad una comparsa tardiva dei sintomi, dopo 1 anno di vita con normale sviluppo psicomotorio.5 In alcuni di questi casi, la sindrome ha un decorso benigno con relativa conservazione del linguaggio e delle capacità motorie. Il nostro paziente con una mutazione nel gene IFIH1 era un caso singolare in quanto aveva anche la sindrome di Singleton-Merten, una malattia rara causata anche da una mutazione nel gene IFIH1 e caratterizzata da displasia dentale, calcificazioni aortiche e osteoporosi.6
il Nostro obiettivo è quello di sottolineare la significativa variabilità fenotipica di AGS e la sua associazione con mutazioni specifiche per lo scopo sia di incoraggiare considerazione questa diagnosi nei casi di presentazioni che si discostano dalla classica forma di malattia e di contribuire ulteriori informazioni sul decorso della malattia e i risultati in questi pazienti.