先天性真皮メラノサイトーシスの異常な提示真皮メラノサイト過誤腫のまれな診断に適合する
編集者へ:
真皮メラノサイトーシスは、胚発生中のメラノブラスト移行の欠陥の結果であると考えられ、真皮に機能的な紡錘形および樹状メラノサイトが存在することを特徴とする。 先天性皮膚メラノサイトーシスは,病変の分布,形態,自然史,および特徴的な組織学的所見に基づいて様々なサブタイプに分類される。 皮膚メラノサイト過誤腫(DMH)の稀なエンティティに合うかもしれない先天性皮膚メラノサイトーシスの異常な症例を提示した。
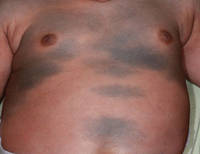
図1. Dermatomal配分に続く鋭い境界ラインが付いている胴上の均一灰色がかった青いパッチ。
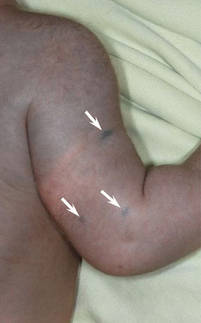
図2. 青みがかったパッチ内の三つの顕著な暗い青色のmacules(矢印)。
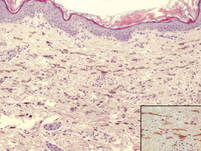
図3. 真皮上部に局在し、皮膚表面に平行に配置された皮内樹状色素メラノサイト(H&E、元の倍率×100)。
4ヶ月の少女は、体幹と上肢に両側の青みがかったパッチを提示しました。 病変は出生以来存在し、18ヶ月のフォローアップ期間中に完全に変化しなかった。 精神的および肉体的発達は正常であった。 色素性障害の家族歴はなかった。 身体検査では、胴体と上肢に均一な灰色がかった青色のパッチが見られた。 パッチは体幹と四肢に沿った皮膚染色体の分布に従っているように見えた(図1)。 いくつかのよく外接された、はるかに暗い青色の黄斑が青みがかったパッチ内に散在していた(図2)。 身体検査の残りの部分は目立たなかった。 完全な血球数および血液化学値は基準範囲内であった。 灰色がかった青色のパッチと顕著な濃い青色の黄斑の両方からの皮膚生検標本は、コラーゲン束の明らかな障害を伴わずに皮膚表面に平行に配置された皮膚双極性樹状色素メラノサイトのかなりの量を示した(図3)。 メラノファージは見られなかった。 臨床所見,組織学的所見,検査所見は先天性真皮メラノサイトーシスの診断と一致した。 基礎となるリソソーム貯蔵疾患は、肝機能検査、腹部超音波検査、および神経学的および眼科的検査を含む代謝スクリーニングによって除外された。
先天性皮膚メラノサイトーシスの臨床スペクトルには、モンゴル斑、太田母斑、伊藤母斑、青色母斑、DMHなどのいくつかの臨床的実体が含まれる。1これらの異なったタイプの皮膚melanocytosis間の微分は特にmelanocytosisのプロセスが広範なとき挑戦的である場合もあります。 先天性皮膚メラノサイトーシスのいくつかのタイプの中で、OtaまたはIto母斑は、臨床的提示に基づいて私たちの患者で除外することができます。
皮膚メラノサイト過誤腫という用語は、BurkhartとGoharaによって導入されました。2それはdermatomalパターンに続く生来の皮膚melanocytosisによって特徴付けられます。 元の症例報告には、私たちの患者に似た灰色がかった青色のパッチの背景を持つ斑点のある暗い青色の黄斑が含まれていました。DMH中の2つのメラノサイトは、典型的には、真皮の下半分に異所性メラノサイトが通常見られる広範なモンゴルの斑点とは対照的に、網状真皮の上半分に位置している。 私たちの患者の色素沈着は、広範なモンゴルのスポット対DMHの診断とより一致しているフォローアップの22ヶ月の間に変更されませんでした。 患者は全身異常を示さなかった。 しかし、先天性メラノサイトーシスおよび神経外胚葉奇形の症例が文献に記載されている。3
真皮メラノサイトーシスの病因についてはほとんど知られていない。 グアニンヌクレオチド結合蛋白質qポリペプチド遺伝子、GNAQ、またはグアニンヌクレオチド結合蛋白質α11遺伝子、GNA11の変異は、マウスの皮膚メラノサイトーシスを引き起こすことが判明した。また、GNAQ変異は、皮膚メラノサイトーシスならびにブドウ膜メラノーマを有するヒトにおいて実証されている。5皮膚メラノサイトーシスの病因の我々の理解の進歩は、皮膚色素沈着障害のより正確な分類につながることが期待されています。