周期性麻痺
臨床的に有用な一次周期性麻痺の分類は、表1に示され、低カリウム血症、高カリウム血症、およびparamyotonic形態を含む。
表1. 一次周期性麻痺(Jurkat-RottおよびLehmann-Hornから修正)(新しいウィンドウでテーブルを開く)
|
病気 |
遺伝子 |
プロテイン |
相続 |
突然変異 |
|
HyperPP |
SCN4A |
Nav1.4 |
ドミナント |
ファイン |
|
ノルモップ |
ファイン(ω-) |
|||
|
パラミョトニアコンゲニータ |
ファイン |
|||
|
HypoPPタイプII |
ファイン(ω-) |
|||
|
へのHypoPP順序 |
CACNA1S |
Cav1.1 |
ドミナント |
ゲイン(ω-) |
|
甲状腺中毒症 |
KCNJ18 |
Kir2.18 |
ドミナント |
損失 |
|
アンデルセン-タウィル症候群 |
KCNJ2 |
Kir2.1 |
ドミナント |
損失 |
弛緩性衰弱の生理学的基礎は、筋肉膜(すなわち、筋膜)の興奮性である。 血清のカリウムのレベルの変化は第一次PPの主な欠陥ではないです;変えられたカリウムの新陳代謝はPPの結果です。 原発性および甲状腺中毒性PPでは,弛緩性麻ひは血清カリウムレベルの比較的小さな変化で起こるが,二次性PPでは血清カリウムレベルが著しく異常である。
このグループの障害には単一のメカニズムは関与していません。 したがって、それらは異質であるが、いくつかの共通の特徴を共有する。 弱さは通常一般化されますが、局在化することがあります。 頭蓋筋および呼吸筋は、通常、免れる。 ストレッチ反射は、攻撃中に存在しないか、または減少しています。 筋線維は、攻撃中に電気的に興奮しない。 筋力は攻撃の間は正常ですが、数年後には、特定のタイプのPP(特にプライマリPP)である程度の固定衰弱が発症します。 原発性PPのすべての形態(becker myotonia congenitaを除く)は、常染色体優性遺伝または散発性(点突然変異から生じる可能性が最も高い)のいずれかである。
電圧に敏感なイオンチャネルは、活動電位の発生(細胞膜の電圧の簡単かつ可逆的な変化)を密接に調節する。 これらは選択的かつ可変透過性のイオンチャネルである。 エネルギー依存性イオン輸送体は濃度勾配を維持する。 活動電位の生成中に,ナトリウムイオンは電圧ゲートイオンチャネルを介して膜を横切って移動する。 安静時の筋線維膜は、主に塩化物チャネルを通る塩化物の動きによって分極され、カリウムの動きによって再分極される。 ナトリウム、塩化物、およびカルシウムチャネル障害は、群として、筋緊張症およびPPと関連している。 ナトリウム、カルシウムおよびカリウムチャネルの機能サブユニットは相同である。 ナトリウムチャネル症はカルシウムや塩化物チャネル症よりもよく理解されている。 すべての形態の家族性PPは、異常な脱分極、ナトリウムチャネルの不活性化、および筋線維の非興奮性を含む最終的なメカニズム経路を示す。
この記事での議論は、主にナトリウム、カルシウム、カリウムチャネルおよびPPの二次形態について説明する。 塩化物チャネル障害は一時的な衰弱と関連しておらず、筋緊張性障害に関する記事でより詳細に議論されている。
様々なタイプのPPにおけるチャネル機能不全の概要
HyperPP高速チャネル不活性化では、変異は通常、膜貫通セグメントの内側部分または細胞内ループに位置し、高速不活性化粒子のドッキング部位に影響を与え、高速チャネル不活性化を損なうため、Na+電流が持続する。
HypoPP過分極活性化カチオン漏れがK+整流電流を相殺すると、変異は最も外側のアルギニンまたはリジン置換を引き起こす。
NormoPP脱分極活性化カチオン漏れにより、コドンR675のドメインIIの電圧センサーのより深い位置に変異がある。
イオンチャネル機能不全は通常、正常な興奮で十分に補償され、持続的な膜脱分極のために筋肉の非興奮性を生成するために追加のトリガーが必要
グルコースとカリウムの摂取は、これらの障害では反対の効果を持っています。 HyperPPでは、カリウムの取入口はブドウ糖がそれを改善する一方攻撃を誘発します。 対照的に、グルコースは低カリウム血症の攻撃を誘発し、カリウムは攻撃の治療法である。
下の画像に注意してください。
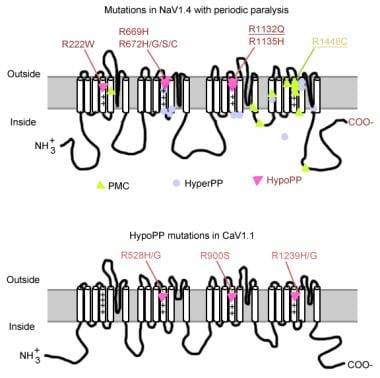
筋肉ナトリウムチャネル遺伝子
ナトリウムチャネルにはαサブユニットとβサブユニットがあります。 ナトリウムチャネルのαサブユニットは、約1800-2000アミノ酸を含む260kd糖タンパク質である。 このチャネルはショウジョウバエからヒトまで進化的に高度に保存されている。 それは4つの相同ドメイン(I-IV)を有し、それぞれが225-325アミノ酸を有する中心細孔を形成する。 各ドメインは、細胞膜を横断する6つの疎水性セグメント(S1-S6)で構成されています。 チャネルの主な機能には、電圧に敏感なゲーティング、不活性化、およびイオン選択性が含まれます。 S5とS6の間の細胞外ループは、原形質膜に浸漬し、細孔の形成に関与しています。 S4セグメントは、すべての第三の位置に正に帯電したアミノ酸を含み、電圧センサとして機能する。 配座の変化は、チャネルの活性化および不活性化をもたらす、脱分極の間に起こり得る。 ドメインIII-S6とドメインIV-S1の間の細胞ループは、不活性化ゲートとして作用する。
ナトリウムチャネルは2つのゲート(活性化と不活性化)を持ち、3つの状態で存在することができる。 分極される膜が付いている残りで活発化のゲートは閉鎖し、不活性化のゲートは開きます。 脱分極により、活性化ゲートが開き、ナトリウムイオンがイオンチャネルを通過することができ、また不活性化ゲートのドッキングサイトが露出する。 脱分極が続くと、不活性化ゲートが閉じ、ナトリウムの細胞への侵入を遮断し、チャネルが急速不活性化状態に入る。 チャネルのこの不活性化は、膜が再分極になることを可能にし、活性化ゲートが閉じ、不活性化ゲートが開いた状態で静止状態に戻る。 二つの不活性化プロセスは、哺乳類の骨格筋に発生します:高速不活性化は、活動電位を終了することを含み、ミリ秒の時間スケールで作用します。 遅い不活性化は数秒から数分かかり、興奮性ナトリウムチャネルの人口を調節することができる。
高速および低速の不活性化を破壊するナトリウムチャネル変異は、通常、HyperPPおよびmyotoniaの表現型と関連しており、低速または高速の不活性化を強化する
ナトリウムチャネル遺伝子(SCN4A)の変異にはいくつかの一般的な特徴があります。 変異のほとんどは、反復IIIとIVの間の”不活性化”リンカー、反復IVの”電圧感知”セグメントS4、または不活性化ゲートのドッキング部位を損なう可能性のある内膜 臨床表現型は特定のアミノ酸置換によって異なり、高カリウム血症PP、paramyotonia congenita(PC)、カリウム増悪筋緊張症(PAM)の間にいくつかの重複が起こることがあるが、3つの表現型は一般的に区別される(後述)。 ほぼすべての変異チャネルは、ナトリウム電流の高速不活性化を障害しています。 ほとんどの患者は全身性カリウムまたは低温に敏感である。
変異型と野生型の二つのチャネル集団が存在し、高速不活性化の障害は変異型筋線維膜の長期脱分極をもたらし、これらの障害の2つの基本的な症状、筋緊張症と衰弱を説明することができる。 高カリウム血症PPでは、機能の利得は過度に影響を受けた筋肉を脱分極する増加されたナトリウムの流れに終って突然変異体チャネルのゲーティング 増加した細胞外カリウム濃度によって引き起こされる可能性がある筋線維膜の軽度の脱分極(5-10mV)は、非活性化モードで維持されている変異チャネ 持続的な内向きナトリウム電流は、剛性(すなわち、筋緊張)として知覚される野生型ナトリウムチャネルの反復焼成を引き起こす。
より重度の脱分極(20-30mV)が存在する場合、正常および異常チャネルの両方が不活性化状態で固定され、衰弱または麻痺を引き起こす。 したがって、膜脱分極の重症度の微妙な違いは、筋緊張症と麻痺の違いを生む可能性があります。 温度の感受性はPCの認刻極印である。 寒さは筋緊張を悪化させ、衰弱を誘発する。 いくつかの突然変異がこの状態に関連しており、そのうち3つはS4セグメントの同じ部位(1448)にある。 これらの変異は、アルギニンを他のアミノ酸に置き換え、この高度に保存されたS4正電荷を中和する。 これらの残基の突然変異は、PCの最も一般的な原因である。 温度感度の原因となる可能性のあるメカニズムには、次のものがあります:
-
温度は変異チャネルの立体配座変化に差動的に影響する可能性がある。
-
より低い温度は異常な状態で変異チャネルを安定化させる可能性がある。
-
突然変異は、リン酸化または第二メッセンジャーなどの他の細胞プロセスに対するチャネルの感受性を変化させる可能性がある。
高カリウム血症PPのほとんどのケースはSCN4A、T704MおよびM1592Vの2つの突然変異が原因です。 ナトリウムチャネルの変異、特に残基1448と1313では、paramyotonia congenitaの原因となっています。 低カリウム血症の周期性麻痺の症例のごく一部は、コドン669および672(Hypopp2)の突然変異と関連している。 Hypopp2では、ナトリウムチャネルの突然変異は機能欠陥の純損失を作り出すために不活性化を高めます。
正常カリウム血症PPはHyperPP(カリウム感受性)とHypoPP(攻撃の持続時間)の両方に似ており、コドン675の電圧センサー DIIのより深い位置でSCN4A変異によって引き起こ R675変異はHypoPPとは異なり、これらの変異は、このサイトが強い脱分極で細胞外サイトにさらされているように逆電圧依存性とω-電流を生成する脱分極活性化ゲーティング細孔をもたらすことである。
カルシウムチャネル遺伝子
カルシウムチャネル遺伝子(CACNL1A3)は、5つのサブユニット(α-1、α-2、β、γ、δ)の複合体である。 骨格筋ジヒドロピリジン(DHP)受容体は、主に横管状膜に位置する。 Α-1サブユニットはDHP薬の結合部位を有し、遅いL型カルシウム電流を伝導する。 それはまた、励起収縮(EC)結合に関与し、筋小胞体(すなわち、カルシウム放出チャネル)のリアノジン受容体との結合を介して電圧センサとして作用する。 膜電位の変化は細胞内カルシウム放出に関連しており、EC結合を可能にする。 DHP受容体/カルシウムチャネルα-1サブユニットの点突然変異は、低カリウム血症PP(Hypopp1)を引き起こす。 CACNA1S遺伝子、R528HおよびR1239Hの二つの突然変異は、低カリウム血症PPのほとんどのケースに責任があります。
疾患の生理学的根拠はまだ理解されていないが、EC結合の失敗ではなく興奮の失敗による可能性が高い。 しかし,低カリウム血症誘発脱分極はカルシウム放出を減少させ,ナトリウムチャネルの不活性化を介してチャネルの電圧制御に直接または間接的に影響を及ぼす可能性がある。 インシュリンおよびアドレナリンは同じような方法で機能するかもしれません。 カルシウムチャネル遺伝子の変異は、SCN4A変異といくつかの類似点を有する。 突然変異はチャネルの不活性化を変更しますが、電圧依存性の活性化は変更しません。 影響を受けた患者からのmyotube培養からの記録は、DHP感受性L型カルシウム電流の30%の減少を明らかにした。 チャネルは低い膜電位で不活性化される。
カルシウムチャネル変異は、電流密度の低下と不活性化の低下として現れる機能の喪失を引き起こす。 この不活性化が低カリウム血症誘発性攻撃とどのように関連しているかは理解されていない。 少なくともR528h変異では、可能な二次channelopathyは、変更されたカルシウム恒常性からATP感受性カリウム電流の減少に関連付けられて発生します。 CACNL1A3変異に関連付けられている低電流はわずかにプロパティとk+チャネル、チャネルの内側整流器クラスに属する特にKATP(ATP感受性カリウムチャ インスリンはまた、この内向き整流器K+電流を減少させることによってHypoPPに作用する。
電圧センサの電荷損失は、HypoPPのほとんどの場合を占めています。 ナトリウムチャネルとカルシウムチャネルは相同な細孔形成アルファサブユニットを有する。 CACNL1A3とSCN4Aの点突然変異は、これらのチャネルのS4電圧センサーのアルゼンチン残基に影響を与えます。 S4セグメントのアルギニン変異は、HypoPPケースの90%を担当しています。
電圧センサの電荷損失は、HypoPPのほとんどの場合を占めています。 ナトリウムチャネルとカルシウムチャネルは相同な細孔形成αサブユニットを有する。 Cav1.1(HypoPP-1)とNav1.4(HypoPP-2)の変異のほとんどすべては、電圧センサーの最も外側のアルギニンまたはリジンのいずれかで正に帯電したアミノ酸を中和します。 Nav1.4つの変異は、最も一般的にはi、II、IIIの繰り返しの電圧センサに位置し、カチオン漏れを引き起こす。
最外アルギニンをグリシンなどのより小さなアミノ酸に置換すると、過分極電位で導電性経路が開き、内部カチオン電流(イオン伝導細孔を介して(ω-)と区別するためのカチオン漏れまたはω電流、s4ゲート細孔を介して一価カチオンの過分極活性化電流であり、整流K+電流を相殺する)が休止電位を脱分極または不安定化する。
s4セグメントは、脱分極中に外側に移動し、導電性経路を閉じる。 重度の電圧センサー変異を有する筋線維は、低カリウム血症の間だけでなく、正常範囲のカリウムレベルでも脱分極され、interictalおよび永久的な衰弱を説明する。 筋肉組織の脂肪置換を伴う重度のミオパチーは、Cav1.1R1239h(DIV変異)の患者に一般的に見られる。
グルココルチコステロイドは、インスリンとアミリンによって媒介されるNa+K+ATPaseを刺激することによってHypoPPを引き起こす。
カリウムチャネル遺伝子
内向き整流はKirチャネルの重要な特性である。 整流には、脱分極中のポリアミンおよびMg++による細孔の電圧依存伝導細孔閉塞が含まれ、この閉塞は過分極中の電位勾配中に除去される。 アンデルセン-タウィル症候群および甲状腺中毒性PPではカリウムチャネル変異が見られる。
異形性特徴、周期性麻痺、および心臓不整脈の三つ組は、アンデルセン-タウィル症候群を特徴付ける。 この症候群は、KCNJ2遺伝子の変異と関連している。 KCNJ2遺伝子は、内向き整流カリウムチャネルKir2.1をコードしています。 KCNE3のカリウムチャネル変異は、低カリウム血症PPを引き起こすことが報告されているが、これは実証されていない。
Kir2.6の変異は甲状腺中毒性PPに対する感受性を引き起こす。 甲状腺中毒性PPで見られるエピソードの弱さはHypoPPおよびAndersen-Tawilシンドロームで見られるそれに類似しています。 この障害は、アジア人とラテンアメリカの男性で最も流行しています。 Thyrotoxic PPはthyrotoxicosisによってマスクされていない遺伝性疾患です。 Kir2.6は主に骨格筋で発現される。 トリヨードチロニンはKcnj18転写を強化し、Kir2.6の発現を増強する可能性があります。 PKCは増加したPIP2転換のためにthyrotoxicosisの間に活動化させ、Kirチャネルは正常なゲーティングの間にPIP2と直接相互に作用します。 Andersen-Tawil症候群では、PIP2親和性が低下しています。 甲状腺中毒性PPでは、変異のどれもKir2.6整流を変更しません。