Aicardi-Goutières症候群:一連の三つの症例における表現型および遺伝的スペクトル|Anales de Pediacía
Aicardi-Goutières症候群(AGS)は、正確な有病率が不明なまれな遺伝性疾患である。 1984年にJean AicardiとFrancoise Goutièresによって、脳脊髄液(CSF)リンパ球増加および基底核の石灰化によって特徴付けられる人生の最初の数ヶ月で発症する進行性脳症とし1それは、過敏性、精神運動遅延、痙性、ジストニア、てんかん発作、無菌性発熱および小頭症の再発エピソードで現れる。 死亡率は脳症期の間に高く、この疾患は典型的にはその後安定するが、重度の神経学的後遺症を引き起こす。 その過程で現れる可能性のある他の特徴は、胆石、眼の症状(主に緑内障)、心臓の関与または自己免疫疾患である。2タイプIインターフェロンは、その発現が増加した生産につながるupregulatedされているAGSの病因に重要な役割を果たしています。3このため、これらの患者における古典的な検査所見の一つは、髄液中のインターフェロンαのレベルの上昇と、pleocytosisおよびneopterinおよびbiopterinのレベルの同等の上昇で 末梢血中のインターフェロンによるインターフェロン刺激遺伝子の発現レベルをマーカーとして評価する潜在的な有用性は、これらのレベルが脳症期(”インターフェロンシグネチャー”)を超えて高いままであるという証拠があるため、現在調査されている。3-5もう一つの重要な特徴は、大脳基底核の石灰化や白質の変化を含む神経イメージング異常の検出である(図。 1). 現在までに、我々は、その変異がインターフェロン経路のアップレギュレーションにつながることができる7つの遺伝子を知っている:ADAR、RNASEH2A、RNASEH2B、RNASEH2C、SAMHD1、TREX1およびIFIH1。 他のすべての遺伝子で報告された変異がホモ接合されているのに対し、ヘテロ接合変異は、TREX1、ADARおよびIFIH1遺伝子について記載されている。IFIH1遺伝子の2つの変異が最近検出された(2014年)4、したがって最も頻度の低い病原性変異であるのに対し、RNASEH2B遺伝子およびTREX1遺伝子の変異は、AGSの診断例の最も高い割合を占めている。
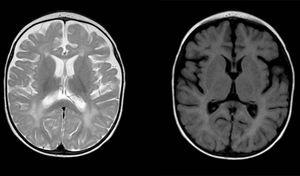
両大脳半球の白質にびまん性および斑状の信号異常、T2重み付け画像では高輝度。 両半球に前頭側頭優位を有するくも膜下腔の拡大、半球間裂の拡大および心室サイズの増加(圧力の増加がない場合)、皮質および皮質下萎縮と適合する。
過去数十年では、これらの特定の突然変異の検出を可能にする遺伝学の進歩のおかげで証拠は原因となる遺伝子に基づく古典的な提示を越える広 我々は、根底にある遺伝的欠陥(表1)に関連して彼らの臨床的特徴を分析することを目的として、過去3年間でAGSの診断を与えられた患者のケースを提示し 一般に、AGSの提示機能は、文献の最新のケースシリーズに記載されているものと一致していた:新生児提示(33%)、小頭症(66%)、精神運動遅延(100%)、痙性(100%)、重度の知的障害(66%)、頭蓋CT上の石灰化(66%)であったが、唯一の患者はてんかん発作を有していた。
Aicardi-Goutières症候群の患者の特徴。
| ケース1 | ケース2 | ケース3 | ||||
|---|---|---|---|---|---|---|
| 遺伝学 | RNASEH2b遺伝子におけるホモ接合変異(P.Ala177Thr) | TREX1遺伝子におけるホモ接合変異(341G>A) | ヘテロ接合変異(c.992C>Gおよびp.IFIH1遺伝子におけるthr331arg) | |||
| 現在の年齢 | 3歳 | 7歳4ヶ月 | 12歳11ヶ月 | |||
| 性別 | 男性 | 女性 | 男性 | |||
| イタリア | ルーマニア | スペイン | イタリア | ルーマニア | イタリア | |
| AP | – | 第36週:子宮内発育制限 第37週: 小頭症、胎盤石灰化 |
口蓋裂 | |||
| 臨床症状 | ||||||
| 発症時年齢 | 10ヶ月 | 出生 | 2歳 | |||
| 最初のプレゼンテーション | 神経過敏 精神運動退行 |
振戦、低血圧、弱い泣き、成長障害 | 運動能力の遅延 | |||
| 精神運動遅延 | はい | はい | はい | |||
| 言語 | 2音節 words | No | Yes | |||
| Microcephaly | Yes | Yes | No | |||
| Chilblains | No | Yes | Yes | |||
| Epilepsy | No | Yes | No | |||
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFCS IV | |||
| 運動障害 | いいえ | はい | いいえ | |||
| 異常な眼球運動 | いいえ | いいえ | いいえ | |||
| 視覚障害 | いいえ | – | 近視 | |||
| 緑内障 | いいえ | いいえ | いいえ | いいえ | ||
| 難聴 | – | – | いいえ | |||
| 心臓の関与 | いいえ | 軽度の三尖弁および僧帽弁逆流 | いいえ | |||
| 再発性発熱 | なし | なし | なし | |||
| 知的障害 | はい | はい、お墓 | はい、軽度 | |||
| その他 | – | – | シングルトン-メルテン症候群: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
|||
| CSF | ||||||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – | |||
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – | |||
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – | |||
| INF alfa (IU/mL) | 25 ( | 75 ( | – | |||
| Blood | ||||||
| Interferon signature | – | – | Gene overexpression | |||
| Neuroimaging | ||||||
| Calcifications on CT | No | Yes. 基底および脳室周囲神経節 | はい。 前頭部およびレンズ状核の深部WMにおける対称石灰化 | |||
| 頭部MRI | 両大脳半球のWM強度のびまん性および斑状の変化、T2上の高輝度。 皮質下WMの関与(U繊維を温存)と脳室周囲WM | 皮質関与なしに、両側および対称的に、前頭葉、側頭葉および後頭葉の皮質下U繊維を含む小葉WMの優位性と一般化されたwm | – | |||
CS F、脳脊髄液;CT、コンピュータ断層撮影;GMFCS、総運動機能分類システム;INF、インターフェロン;IUGR、子宮内増殖制限;MRI、磁気共鳴画像法;PNP、多発神経障害;WM、white mater.
前述のように、RNASEH2b遺伝子におけるホモ接合変異は、AGSを引き起こす最も頻繁な変異体であり、その表現型発現は、通常、古典的な提示に最も適合する。4これは、過敏性および精神運動遅延および特徴的な神経画像およびCSF所見を有する10ヶ月齢で発症したこのような変異を有する患者の症例であった。
AGSの症例の20%が新生児の提示を示し、子宮内で発症する疾患が発症する可能性がある。前述の7つの遺伝子のいずれかにおける5つの突然変異は、この表現型につながる可能性があるが、この初期の提示は、最も頻繁にTREX遺伝子と関連して4,5この形態の最初の提示はトーチの伝染のそれに類似しています、hepatosplenomegaly、hypertransaminasaemia、血小板減少症および極度な過敏症、運動障害およびepileptic捕捉を含む神経学的な明示と。5これらの患者に病気のより厳しいコースがあり、死のより高い危険にあります。 このような変異体を提示した私たちのサンプルの患者は、新生児の提示を有し、現在3の最も重篤な形態の病気を有する。
ADAR1遺伝子、特にIFIH1遺伝子の変異は、正常な精神運動発達を伴う1年間の生活の後、症状の遅発性と関連している。これらの症例のうちのいくつかでは、この症候群は、言語および運動能力の相対的な保存を伴う良性の経過を有する。 IFIH1遺伝子の変異を持つ私たちの患者は、彼はまた、シングルトン-メルテン症候群、またIFIH1遺伝子の変異によって引き起こされ、歯科異形成、大動脈石灰化6
私たちの目的は、AGSの重要な表現型の変動性と特定の変異との関連を強調することで、古典的な疾患の形態から逸脱した提示を有する症例におけるこの診断の検討を奨励し、これらの患者の疾患および転帰に関する追加情報を提供することを目的としている。