ATrain Education
パーキンソン病の病態生理について毎日学んでいますが、依然として主に特発性(原因不明)と考えられています。 それはおそらく宿主感受性と環境要因の相互作用を含む。 症例のわずかな割合は遺伝的に関連しており、遺伝的要因が激しく研究されている。
生理学的には、パーキンソン病に関連する症状は、多くの神経伝達物質、最も顕著なドーパミンの喪失の結果である。 病気によって影響される細胞の多くが失われると同時に徴候はそのうちに悪化します。 病気の経過は非常に可変であり、年齢とともに非常に症状が少なく、症状が急速に進行する患者もいます。
パーキンソン病は、一連の進行を伴う複雑な神経変性疾患としてますます認識されています。 それが最初に迷走神経の背側運動核および嗅覚の球根および核、そして軌跡のcoeruleus、および最終的に黒質に影響を与えるという強い証拠があります。 脳の皮質領域は後の段階で影響を受ける。 これらの様々な神経系への損傷は、運動系だけでなく、認知および神経心理学的系にも障害を引き起こす多面的な病態生理学的変化を説明する(Kwan&Whitehill,2011)。
ドーパミンの役割
ドーパミンは、他の神経伝達物質と同様に、シナプス(シナプス前細胞とシナプス後受容体の間の空間)を横切って、ある神経細胞か ドーパミンはシナプス前膜の膜貯蔵小胞からシナプスに分泌される。 それはシナプスを交差させ、ドーパミン受容体を活性化するシナプス後膜に結合する。 シナプスに残っている未使用のドーパミンはシナプス前細胞に戻って吸収され、シナプス前細胞に戻ったら、過剰なドーパミンは貯蔵小胞に再パッケージ化され、シナプスに再び放出される。
シナプス内では、ドーパミンがある細胞から別の細胞に移動すると、MAO(monoamine oxidase)とCOMT(catechol-O-methyl transferase)の二つの酵素によって分解され、不活性になることがあります。 一つの治療戦略は、MAO酵素の作用を中断し、ドーパミンの分解を防止するシナプスにMAO阻害剤を導入する。 これはより多くのドーパミンがシナプスに残るようにし、postsynaptic膜に結合する可能性を高めます。
化学シナプス伝達
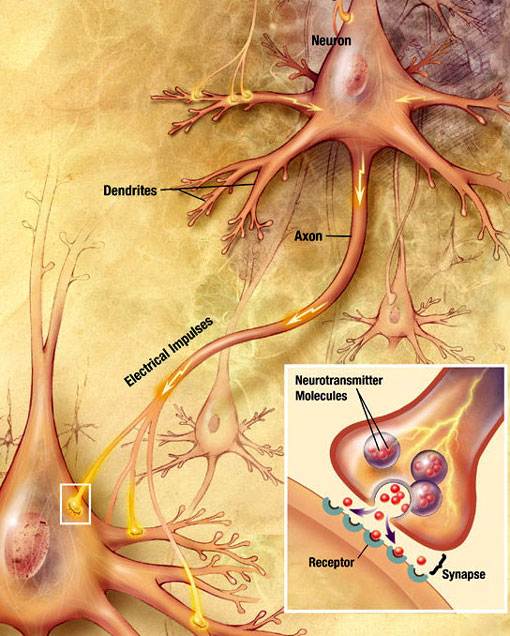
活動電位と呼ばれる電気化学的な波がニューロンの軸索に沿って移動する。 活動電位がシナプス前末端に達すると、シナプス後ニューロンの膜に位置する化学受容体分子に結合する少量の神経伝達物質分子の放出を誘発し、シナプス裂け目の反対側にある。 出典:ウィキメディア-コモンズ。
正常およびパーキンソン病に罹患したニューロンにおけるドーパミンレベルの進行性の喪失
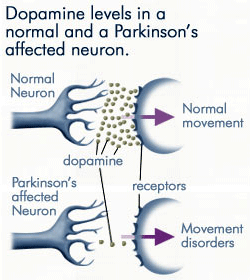
パーキンソン病の影響を受けるニューロンによってドーパミンが産生されることが少なくなるにつれて、シナプス後膜上のドーパミン受容体に結合する ソース:anti-agingfirewalls.com.
ドーパミン細胞の損失を直接測定することはできませんが、神経学的に正常な人々と非ヒト霊長類の測定は、年齢とともにドーパミンの遅い進行性の損失を明らかにする。 パーキンソン病で損失は大いにより大きい率に起こり、生化学的な手段およびイメージ投射調査は両方運動徴候が現われる時間までにドーパミンに重要な減 この見解では、パーキンソン病は、正常な老化に伴って見られる細胞死の加速バージョンである(Cookson、2009)。 これは、通常の老化、特発性PD、環境または遺伝的要因によって引き起こされるPD、および早期発症PDにおけるドーパミン作動性ニューロンの減少を示す下のグラフに示されている。
パーキンソン病におけるドーパミン枯渇の進化
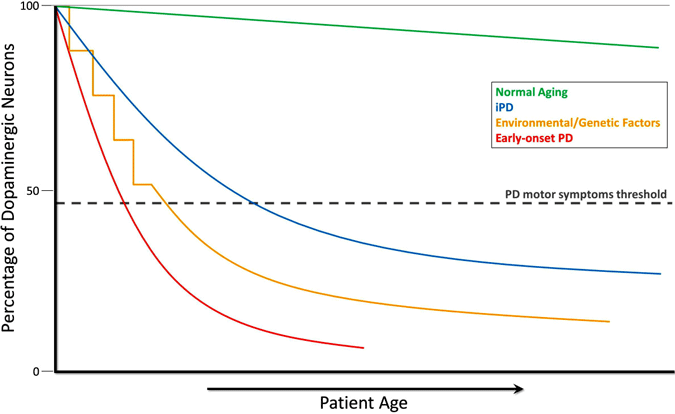
通常の加齢中(緑色の線)、小さいが遅いドーパミン変性は運動症状なしに起こる。 特発性PD(IPD、ブルーライン)は未知の起源であるが、徐々に発達すると考えられており、ドーパミン作動性ニューロンのゆっくりとした変性が、後の人生で古典的なPD運動症状をもたらすと考えられている。 PD運動症状につながるドーパミン神経変性の別のモデルは、ドーパミン作動性ニューロン損失(黄色線)への遺伝的素因と組み合わせて、時間をかけて環境毒 パーキン遺伝子の変異によって引き起こされる早期発症PD(赤線)は、ドーパミン作動性ニューロンの急激な低下を伴い、PD運動症状は特発性PDのものよりも数十年前に現れる可能性がある。 PD運動症状発症のもう1つのシナリオ(図示せず)は、出生時に非典型的に低い数のドーパミン作動性ニューロンをもたらし、PD発症に対する感受性を増, 2012).
ドーパミンニューロンの変性は、pars compactaと呼ばれる黒質の一部で特に顕著である。 有意に、pars compactaにおけるドーパミンの損失は、大脳基底核における全体的な興奮性駆動を増加させ、自発的な運動制御を破壊し、PDの特徴的な症状を引き起こ 運動機能の正常化は、レボドパ処理で最初に見られる(Gasparini e t a l., 2013).
*大脳基底核の主要な構成要素は、線条体(尾状核および被殻)、淡蒼球、黒質、側坐核および視床下核である。
PDの重症度が高まるにつれて、ドーパミンの枯渇は、グルタミン酸、GABA、セロトニンなどの他の基底核神経伝達物質の機能の変化を含む、基底核経路のさらなる変化をもたらす(Gasparini et al., 2013). 黒質にはドーパミン産生ニューロンの相対的な脆弱性がありますが、すべてのドーパミン細胞がパーキンソン病に罹患しているわけではありません。
黒質線条体経路
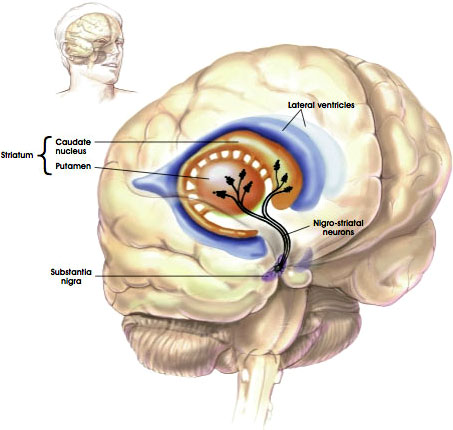
出典:NIH,n.d.
レビー小体およびΑ-シヌクレイン
レビー小体は、パーキンソン病患者の神経細胞内で発生する異常な凝集体およびタンパク質の封入体である。 凝集体は、通常、ミスフォールドされたタンパク質を含む不溶性線維性凝集体からなる。 レビー体では多数の分子が同定されているが、α-シヌクレインと呼ばれるタンパク質が主成分である。
レビー小体(Α-シヌクレイン封入体)
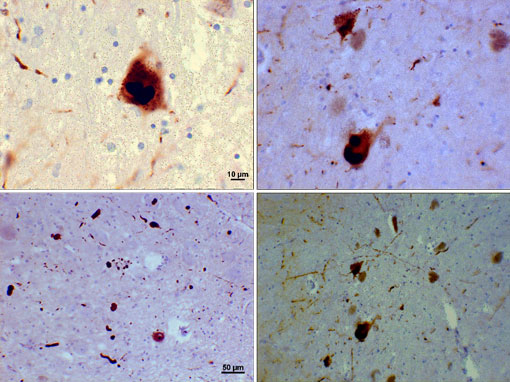
様々な倍率でレビー体とレビー神経突起を示すパーキンソン病患者の黒質の領域の顕微鏡写真。 上部パネルは、レビー体を形成するために凝集したα-シヌクレイン内膜介在物の60倍の倍率を示す。 下部パネルは、様々なサイズのストランド様レビー神経突起と丸みを帯びたレビー体を示す20倍の倍率の画像です。 Surajラージャンの画像の礼儀。
レビー病理学は脳の多くの領域を包含し、いくつかの報告は、黒質がパーキンソン病でレビー体が形成される最初の場所ではないことを示唆している。 インクルージョンと凝集体は、複雑なイベントのカスケードの終わりの段階を象徴する可能性があります。 初期の段階は、診断的特徴を表す場合と表さない場合がある介在物自体よりも、障害の病因に直接結びついている可能性があります。
レビー小体は「レビー小体を伴う痴呆」にも見られ、これらの状態は共通の病理学によって、そしておそらく共通の病因によって互いに関連していることを示唆している。 細胞損失もレビー体の形成もPDには絶対的に特異的ではないが、両方とも現在の定義の下でPDの診断に必要である(Cookson、2009)。
アルツハイマー病、前頭側頭変性症、プリオン病、ハンチントン舞踏病、運動ニューロン病などの神経変性疾患は、神経系の特定の領域におけるタンパク質の凝集や封入体の形成など、共通の細胞および分子メカニズムを持つことがますます実現されている(Jellinger,2011)。
炎症と免疫応答
ドーパミン作動性変性の引き金は、内因性要素と環境要素の両方によって多因子的に影響されるようです。 炎症および免疫応答は、ドーパミン作動性変性の重要なメディエーターとしてますます考慮されている。 大規模な集団研究は、非ステロイド性抗炎症薬(Nsaid)を服用している個人が特発性PDを発症するリスクが低いことを示唆しており、抗炎症薬がパーキンソ
新しい試験段階では、抗炎症治療が関与しており、特にPD患者の炎症性変化を減少させることを目的とした治療における客観的なバイオマーカーを探 研究者は、大規模な臨床画像試験でこれをテストすることを意図して、関連するバイオマーカーを開発するために神経画像ツールを使用しています。 これらの試験の結果は、PDの抗炎症治療の進行をテストし、監視するためのデータを提供し、停止する、または少なくとも遅い、炎症性媒介ドーパミン作動性変性(Barcia、2013)
パーキンソニズム
パーキンソニズムは、”非定型パーキンソン病”、”続発性パーキンソン病”、または”パーキンソン症候群”としても知られており、患者がパーキンソン病に関連する症状のいくつか—振戦、剛性、運動緩慢、および姿勢不安定性を示す神経学的症候群である。 しかし、パーキンソン病はパーキンソン病ではありません。 パーキンソニズムはパーキンソン病によって引き起こされると考えられないし、患者はpharmacologic介在に普通不完全に答えます。 パーキンソニズムは、多くの場合、毒素、メタンフェタミン、外傷、複数の脳卒中、他の神経系障害、または病気への暴露などの識別可能な原因を持っています。 一般的に、レビー体はパーキンソニズムでは見られない。
パーキンソニズムという用語は、進行性核上麻痺、多系統萎縮症、レビー体痴呆、皮質基底変性、血管パーキンソニズム、薬物誘発パーキンソニズム、および感染および他の原因に続発するパーキンソニズムなどの障害にも関連している(Hohler et al., 2012). 可逆的パーキンソニズムの一形態は、特定の神経弛緩薬、特にレセルピン、抗精神病薬(ハロペリドール)、およびメトクロプラミドの使用から起こり得る。 特定の毒素への暴露、重度の一酸化炭素中毒、および水銀中毒もパーキンソニズムにつながる可能性があります。
1980年代初頭、合成アヘン剤の汚染されたバッチを消費した薬物中毒者のグループにおけるパーキンソニズム症状の出現は、非ヒト霊長類およびヒトにおいてパーキンソニズム症候群を引き起こす薬剤としての化学MPTPの発見につながった。 MPTPはヘロインの形態を作るとき作り出すことができます(MPTPは選択的に黒質のドーパミンの細胞を破壊する神経毒に変えられます)。 これらの症例はまれであり、主に長期的な薬物使用者に影響を与えている。
メタンフェタミンの乱用もパーキンソニズムと関連している。 実験動物では、メタンフェタミンへの暴露は、線条体*のドーパミン作動性繊維だけでなく、黒質の細胞体を損傷し、PD患者で観察された変性をエコーする。 線条体中のドーパミン作動性末端への選択的損傷は、ヒトのメタンフェタミン使用者においても観察されているが、メタンフェタミン乱用が黒質中のドーパミン作動性細胞体に損傷を与えるという証拠はこれまでにない(Granado et al., 2013).
*大脳基底核の最大の核であり、線条体は尾状核と被殻からなる。
メタンフェタミンの使用は、ユーザーに将来のPDの開発の素因となる可能性があるという仮説が立てられています。 この仮説はメタンフェタミンのユーザーにPDを開発する高められた危険があることを示す最近の疫学的な仕事によって支えられました。 これは、実験動物におけるメタンフェタミンの持続的な神経毒性効果と一致する(Granado e t a l., 2013).
パーキンソニズムの患者は、外来患者として管理することが困難なことが多い。 それらの症状の複雑さ、追加された認知および自律神経の欠損、ほとんどのPD薬に対する反応不良、および状態の比較的急速な低下は、特に疾患が進行, 2012).