Aicardi-Goutiè syndrom: Fenotypisk og genetisk spektrum I en serie Av tre tilfeller | Anales De Pediatrí
Aicardi-Goutiè syndrom (AGS) er en sjelden arvelig sykdom med eksakt prevalens ukjent. Det ble først beskrevet i 1984 Av Jean Aicardi Og Francoise Goutiè som en progressiv encefalopati med utbrudd i de første månedene av livet preget av cerebrospinalvæske (CSF) lymfocytose og forkalkninger i basalgangliene.1 det manifesterer seg med irritabilitet, psykomotorisk retardasjon, spastisitet, dystoni, epileptiske anfall, tilbakevendende episoder av aseptisk feber og mikrocefali. Dødeligheten er høyere i den encefalopatiske fasen, og selv om sykdommen vanligvis stabiliserer seg etterpå, forårsaker den alvorlige nevrologiske følger. Andre karakteristiske trekk som kan oppstå i løpet av kurset er chilblains, okulære symptomer (hovedsakelig glaukom), hjerte involvering eller autoimmune lidelser.2 type i interferoner spiller en avgjørende rolle I patogenesen AV AGS, hvor deres uttrykk er oppregulert, noe som fører til økt produksjon.3 av denne grunn er et av de klassiske laboratoriefunnene hos disse pasientene et forhøyet nivå av interferon alfa i CSF, sammen med pleocytose og like forhøyede nivåer av neopterin og biopterin. Den potensielle nytten av å vurdere nivået av ekspresjon av interferonstimulerte gener av interferon i perifert blod som markør undersøkes for tiden, da det er tegn på at disse nivåene holder seg høye forbi den encefalopatiske fasen («interferon signatur»).3-5 en annen viktig funksjon er påvisning av neuroimaging abnormiteter inkludert forkalkninger i basalganglia og endringer i den hvite saken (Fig. 1). Til dags dato vet vi om 7 gener hvis mutasjoner kan føre TIL oppregulering av interferonveien: ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1 og IFIH1. Heterozygote mutasjoner har blitt beskrevet FOR trex1, ADAR og IFIH1 gener, mens mutasjonene rapportert i alle andre gener har vært homozygote.2 Mutasjoner i IFIH1-genet ble påvist sist (2014) 4 og er derfor de minst hyppige patogene variantene, mens mutasjoner I rnaseh2b-og TREX1-genene står for den høyeste andelen diagnostiserte TILFELLER AV AGS.
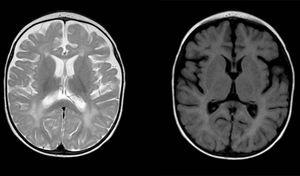
Diffuse og usammenhengende signalavvik i hvit substans i begge hjernehalvdelene, hyperintense i t2-vektede bilder. Forstørret subaraknoidal plass med frontotemporal overvekt i begge halvkuler, med utvidelse av interhemispheric fissur og økt ventrikulær størrelse (i fravær av økt trykk), kompatibel med kortikal og subkortisk atrofi.
I de siste tiårene, takket være fremskritt innen genetikk som tillater påvisning av disse spesifikke mutasjonene, har det oppstått et bredt fenotypisk spektrum utover den klassiske presentasjonen basert på det forårsakende genet. Vi presenterer tilfellene av 3 pasienter som har fått DIAGNOSEN AGS de siste 8 årene med sikte på å analysere deres kliniske egenskaper i forhold til den underliggende genetiske defekten (Tabell 1). GENERELT var presentasjonsegenskapene til AGS i samsvar med de som er beskrevet i den siste kasuserien i litteraturen: neonatal presentasjon (33%), mikrocefali (66%), psykomotorisk retardasjon (100%), spastisitet (100%), alvorlig intellektuell funksjonshemning (66%) og forkalkninger på kranial CT (66%), selv om bare en pasient hadde epileptiske anfall.
Egenskaper hos pasienter med aicardi-Goutiè syndrom.
| Sak 1 | Sak 2 | Sak 3 | |
|---|---|---|---|
| Genetikk | Homozygot mutasjon (S. Ala177Thr) I rnaseh2b-genet | Homozygot mutasjon (341g > A) I trex1-genet | Heterozygot mutasjon (c.992c> G OG p.Thr331Arg) I IFIH1 genet |
| Nåværende alder | 3 år | 7 år og 4 måneder | 12 år og 11 måneder |
| Kjønn | Hann | Hunn | Hann |
| Opprinnelse | Romania | Spania | Italia |
| AP | – | Uke 36: intrauterin vekstrestriksjon Uke 37: mikrocefali, kalsifisering av placenta |
Ganespalte |
| Kliniske manifestasjoner | |||
| alder ved debut | 10 måneder | Fødsel | 2 år |
| Innledende presentasjon | Irritabilitet Psykomotorisk regresjon |
Skjelvinger, hypotoni, svakt gråt ,vekstsvikt | Motorisk forsinkelse |
| Psykomotorisk retardasjon | Ja | Ja | Ja |
| Språk | 2-Stavelse words | No | Yes |
| Microcephaly | Yes | Yes | No |
| Chilblains | No | Yes | Yes |
| Epilepsy | No | Yes | No |
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFCS IV |
| bevegelsesforstyrrelser | Nei | Ja | Nei |
| Unormale øyebevegelser | Nei | Nei | Nei |
| synshemming | Nei | – | Myopi |
| Glaukom | Nei | Nei | Nei |
| Hørselstap | – | – | Nei |
| hjertesykdom | Nei | Mild trikuspidal og mitralregurgitasjon | Nei |
| Tilbakevendende feber | Nei | Nei | Nei |
| Utviklingshemming | Ja | ja, grav | ja, mild |
| Andre | – | – | Singleton-Merten syndrom: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
| CSF | |||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – |
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – |
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – |
| INF alfa (IU/mL) | 25 ( | 75 ( | – |
| Blood | |||
| Interferon signature | – | – | Gene overexpression |
| Neuroimaging | |||
| Calcifications on CT | No | Yes. I basale og periventrikulære ganglier | Ja. Symmetriske forkalkninger i dyp WM av frontal region og lentiform nucleus |
| HODE MRI | Diffuse og usammenhengende endringer I WM intensitet av begge hjernehalvdelene, hyperintense På T2. Involvering av subkortisk WM (sparsom U-fibrene) og periventrikulær wm | Generalisert wm-involvering med overvekt av lobar wm inkludert de subkortiske U-fibrene i de frontale, temporale og occipitale lobene, bilateralt og symmetrisk, uten kortikal involvering | – |
CSF, cerebrospinalvæske; CT, computertomografi; GMFCS, Grovmotorisk Funksjon Klassifiseringssystem; INF, interferon; iugr, intrauterin vekstbegrensning; MR, magnetisk resonansavbildning; PNP, polyneuropati; WM, hvit mater.
som nevnt tidligere er homozygote mutasjoner I rnaseh2b-genet de hyppigste varianter som forårsaker AGS, og deres fenotypiske uttrykk samsvarer vanligvis mest med den klassiske presentasjonen.4 dette var tilfellet for pasienten i vår studie som hadde en slik mutasjon, som hadde oppstått i en alder av 10 måneder med irritabilitet og psykomotorisk retardasjon og med karakteristiske neuroimaging og CSF-funn.
Tjue prosent av TILFELLENE AV AGS kan ha en nyfødt presentasjon, med sykdomsutbrudd i utero.5 Mutasjoner i noen av de 7 nevnte gener kan føre til denne fenotypen, men denne tidlige presentasjonen er oftest assosiert MED trex-genet.4,5 den første presentasjonen av dette skjemaet ligner PÅ EN TORCH-infeksjon, med hepatosplenomegali, hypertransaminasemi, trombocytopeni og nevrologiske manifestasjoner, inkludert ekstrem irritabilitet, bevegelsesforstyrrelser og epileptiske anfall.5 disse pasientene har en mer alvorlig sykdomssykdom og har høyere risiko for død. Pasienten i vår prøve som presenterte med en slik variant hadde en neonatal presentasjon og har for tiden den mest alvorlige sykdomsformen til 3.
Mutasjoner I adar1-genet og SPESIELT IFIH1-genet er forbundet med en sen symptomstart, etter 1 års levetid med normal psykomotorisk utvikling.5 i noen av disse tilfellene har syndromet et godartet kurs med relativ bevaring av språk og motoriske ferdigheter. Vår pasient med en mutasjon i IFIH1 genet var en entall i at han også hadde Singleton-Merten syndrom, en sjelden sykdom også forårsaket av en mutasjon I IFIH1 genet og preget av dental dysplasi, aorta forkalkninger og osteoporose.6
vårt mål er å understreke den signifikante fenotypiske variabiliteten AV AGS og dens tilknytning til spesifikke mutasjoner med det formål å både oppmuntre til vurdering av denne diagnosen i tilfeller med presentasjoner som avviker fra den klassiske sykdomsformen og å bidra med ytterligere informasjon om sykdomsforløp og utfall hos disse pasientene.