Gynekologi & Obstetrikk Saksrapport
Nøkkelord
Tetra-amelia; Misdannelse; Genotype; Fenotype.
Innledning
Lem anomalier utgjør en viktig gruppe medfødte patologier preget av hypoplasi eller fullstendig aplasi av en eller flere lemben. Lem abnormiteter av alle typer forekommer i ca 1 av 1300 til 2000 fødsler. Disse lemavvikene kan isoleres eller knyttes til andre anomalier . Tetraamelia syndrom er sjelden og grå områder forblir.
vi rapporterer to tilfeller av tetra-amelia i et NIVÅ II fødselspermisjon I Dakar (Senegal) med å være lik tetraamelia-1 (kromosom 17q21), tetraamelia-2 (kromosom 8q23) Og Roberts syndrom (kromosom 8p21). Dette illustrerer vanskeligheten med å korrelere fenotype og gener involvert.
Saksrapporter
Sak 1
MS AD var en 44 år gammel mor henvist til vår avdeling ved 36 ukers svangerskap med alvorlige preeklampsi og føtale anomalier. Hun var fem para uten historie av foster anomalier. Hun røykte ikke nå og hadde aldri røykt eller drukket alkohol. Hun hadde ikke vært utsatt for passiv røyking. Hun var i en tredje grad consanguineous ekteskap for alle sine barn. MS. AD hadde testet negativt for hepatitt B, HIV og syfilis. Hun var beskyttet mot rubella-viruset og hadde ingen tidligere eksponering for Toxoplasma gondii. Ultralyd overvåking utført sent på 33 uker og 35 uker av svangerskapet hentet oligoamniosis og hydrocephalus samt agenesis av lemmer. Resept under graviditet inkluderte administrering av jern og folsyre, samt administrering av sulfadoksinpyrimetamin. Sistnevnte ble foreskrevet ved 18 uker og deretter 26 uker som en del av anti-malaria profylakse politikk for gravide kvinner. Symphyseal-fundal høyde målt 28 cm. På grunn av alvorlige trekk ved preeklampsi; hun ble innlagt på sykehus umiddelbart og observert i en arbeids-og leveringsenhet. Hun fikk DA først iv magnesiumsulfat FOR å forhindre eclampsia og antihypertensive medisiner for å opprettholde systolisk blodtrykk under 160 mmHg og diastolisk blodtrykk under 105 mmHg.
beslutningen om umiddelbar keisersnitt ble gjort. En 2,150 gram levende født ble ekstrahert som senere døde innen 10 minutter. Før døden presenterte kroppen krypende bevegelser. Flere eksterne anomalier ble identifisert (Figur 1) inkludert komplett agenese av alle fire lemmer, hydrocephalus med en hodeomkrets på 39 cm. På ansiktet var det hypertelorisme med et kolobom i øyelokkene, mild exophthalmos og aniridia. Munnen var lik en omvendt V uten klar avgrensning av leppene og nesen var rudimentær. Det nyfødte var blottet for integrasjoner(hår og øyenbryn). Ørene ble redusert til skisser som så ut som slisser. Nakken var kort. Stammen ble redusert til en 26 cm lang konisk struktur med en navlestreng i bunnen. Brystet var smalt. Like under navlen, stammen utvidet bakover var til stede på gulvet i halepol, en midtlinje med resesjoner og en spirende som kan tilsvare en fallos av ubestemt type. Agenesis av bekkenet, kjønnsorganer og anal imperforation ble notert. Fosterpatologi har ikke blitt utført. Imidlertid kan død innen 10 minutter etter levering og det koniske utseendet på brystet foreslå lungeavvik.
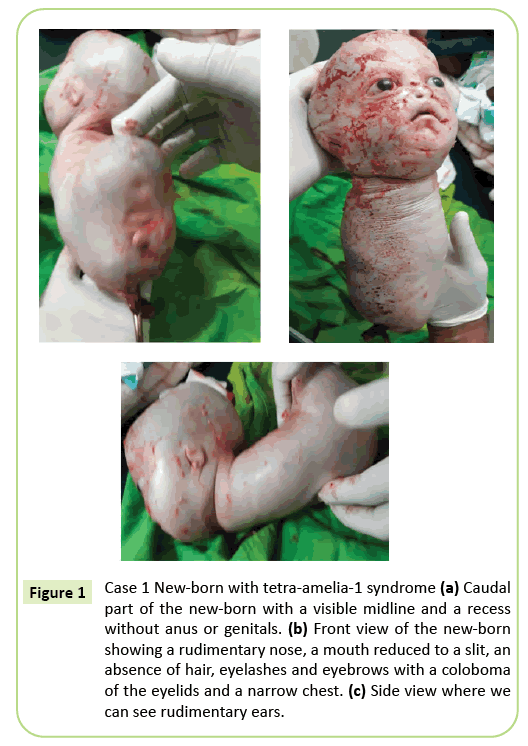
Figur 1: Tilfelle 1 Nyfødt med tetra-amelia-1 syndrom (a) Caudal del av den nyfødte med en synlig midtlinje og en utsparing uten anus eller kjønnsorganer. (b) forfra av den nyfødte viser en rudimentær nese, en munn redusert til en slit, et fravær av hår, øyevipper og bryn med en coloboma av øyelokkene og en smal bryst. (c) Sidevisning hvor vi kan se rudimentære ører.
Sak 2
det andre tilfellet var en 22-årig primigravida henvist til vårt anlegg for ultralydsskanning ved 37 ukers svangerskap. Hun var ikke i en consanguineous ekteskap. Hun hadde testet negativt for hepatitt B, HIV og syfilis. Hun ble ikke testet For Toxoplasmose og rubella. Ingen ultralyd overvåking ble gjort i løpet av svangerskapet. Den kliniske undersøkelsen var i samsvar med føtal vekstretardasjon(Fundal høyde: 26 cm). Ultralydfunn viste at humerus ble forvrengt som måler 23, 9 mm tilsvarende 17 ukers svangerskap. Det var agenesis av lårbenet. Iliac vingene var synlige på ultralyd. Ingen lunge – eller hjerteavvik ble identifisert. Levering ble påbegynt. Den nyfødte hadde en kvinnelig fenotype Med En Apgar score på 9 ved 5. minutt. Morfologien til hodet og stammen var uten særpreg. Øvre lemmer ble redusert til to 3 cm lange stubber. Komplett agenese av de 2 nedre lemmer ble notert. Det var en symmetrisk anomali(Figur 2).
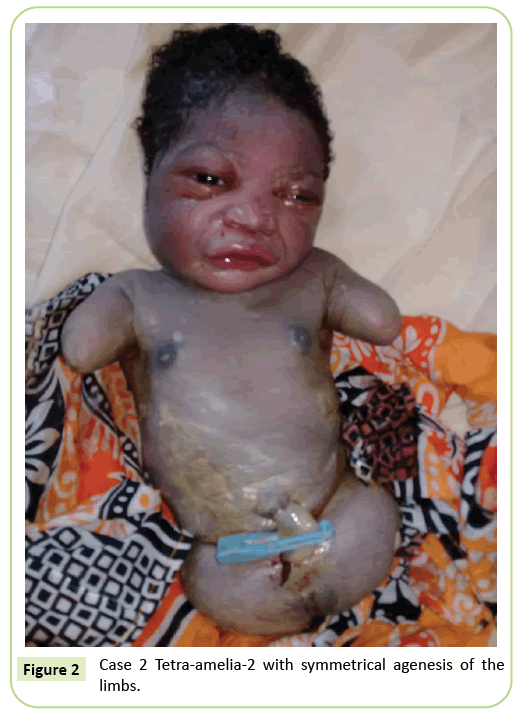
Figur 2: Sak 2 Tetra-amelia-2 med symmetrisk agenese av lemmer.
Diskusjon
Bermejo-Sanchez et al. beskrevet i 2011 epidemiologi av medfødt amelia ved hjelp av data samlet inn fra 20 medfødte anomalier overvåkingsprogrammer, fra alle kontinenter unntatt Afrika, mellom 1968 og 2006. Totalt ble 326 tilfeller av amelia identifisert blant 23.110.591 levendefødte, dødfødsler og aborter. Prevalensen var 1.41 / 100.000 .
Tetra-amelia refererer til som fullstendig fravær av lemmer og forekommer sjelden. Etter vår kunnskap er tetra-amelia-1 beskrevet i 7 familier. Det ser ut til å følge en autosomal recessiv arv. I alle familier var tetra-amelia-1 forbundet med alvorlige misdannelser i andre deler av kroppen, inkludert ansikt og hode, anomalier i nervesystemet, skjelett og kjønnsorganer. Lungene var underutviklet i mange tilfeller, noe som gjør pusten vanskelig eller umulig . Zimmer et al. rapportert i 1985 en sterkt innavlet familie der 6 spedbarn hadde tetra-amelia-1 og hydrocephalus. De beskrev i et av fostrene et totalt fravær av bekkenbenet, spalt leppe og gane, arrhinia og aplasi i ørene. En bilateral venstre lunge, en vedvarende arteriell kanal, en anal imperforasjon ble også notert. Fostertesting eliminerte diagnosen Roberts syndrom . Andre tilfeller hentet i litteraturen er At Av Kosaki et al., i 1996, med et foster av karyotype 46, XX med tetraphocomelia og alvorlig lungehypoplasi i tillegg til ansikts-og hodeanomalier . Rosenak et al. beskrevet et tilfelle av tetra-amelia med alvorlig lungehypoplasi i to foster av et ikke-consanguineous par. Fostertesting utelukket diagnose Av Roberts syndrom . Ytterligere to tilfeller ble rapportert Av Zlotogora et al. i 1993. Begge pasientene døde kort tid etter fødselen, og forfatterne foreslo eksistensen av lungehypoplasi. Niemann et al. rapportert en consanguineous tyrkisk familie der 4 av de 8 brødrene led av tetra-amelia. I tillegg til fraværet av de 4 lemmer, viste fosterundersøkelsene av 3 foster flere anomalier: spalte lepper og /eller palatin, laparoschisis, lungeanomalier, bekkenhypoplasi, atresi av choanas, vagina og anal imperforasjon . Til slutt, I 2005, Krahn et al. beskrevet 2 brødre født av innavlede foreldre som lider av tetraamelia og alvorlig lungehypoplasi. Kragebenene og skulderbladene var normale i det andre fosteret. Karyotypen var normal .
Tetra-amelia-1 syndrom eller TETAMS1 er forårsaket av en homozygot mutasjon I wnt3 genet på kromosom 17q21 med en autosomal recessiv arv. Tetraamelia-2 syndrom (TETAMS2) er preget av rudimentære lemmer eller et komplett fravær av lemmer, generelt symmetrisk samt bilateral agenese av lungene i noen tilfeller. Er også vanlige anomalier i det pulmonale vaskulære systemet og dysmorfier inkludert bilateral leppe-og ganespalte, ankyloglossi, mandibulær hypoplasi, mikroretrognati og labioskrotal aplasi .
Szenker-Ravi, som studerte 4 familier av tetra-amelia med agenesis eller lungehypoplasi, bemerket en fenotypisk heterogenitet med lem anomalier av varierende alvorlighetsgrad . Eksomsekvensering i disse 4 familiene har gjort det mulig å identifisere avkortende homozygote mutasjoner I rspo2-genet . Tetraamelia-2 syndrom er forårsaket av en homozygot mutasjon I rspo2-genet (610575) som ligger på kromosom 8q23 .
fenotypen til det første tilfellet beskrevet i denne artikkelen tilsvarer et tetra-amelia-1 syndrom på grunn av spesielt tilstedeværelsen av hydrocephalus, anomalier kjønnsorganer og en rudimentær nese. Den smale brystet og tidlig død før 10. minutt av livet tyder på alvorlig lungehypoplasi. Denne saken fremhever fenotypisk heterogenitet med øyelokkkolobom, hypertelorisme, eksofthalmos og sjeldne vedlegg.
vi anser det andre tilfellet i vår studie for å være et tetramelia-2 syndrom med tanke på symmetrisk tetra-amelia med tilstedeværelse av øvre lemstubber. Diagnose av tetra-amelia bør gjøres tidlig under ultralydovervåking. Derfor bør man øke bevisstheten om viktigheten av ultralydovervåking og bruk AV 3D / 4D for å forbedre screeningsresultatene. Diagnosen av en bekkenmasse på ultralyd parret med amelia bør øke mistanke om spleno-gonadal fusjon lem defekt syndrom.
i tillegg skal fosterets undersøkelse og føtal testing ved hjelp av utviklende teknologier av kromosomal microarray og exome og genome sekvensering oppfordres i våre innstillinger. En bedre karakterisering av tilfellene gjør det mulig å gi råd til par og bedre kunnskap om disse kliniske anomaliene.
Konklusjon
Tetra-amelia syndrom er knappe Og grå områder fortsatt. Disse to tilfellene, sammenlignet med det som allerede er beskrevet i litteraturen, illustrerer tetraameliens fenotypiske heterogenitet. Gitt den sjeldne forekomsten av disse anomaliene, ville det være viktig å opprette et internasjonalt register over anomalier for å rapportere tilfeller og sette opp en prøvebank for utvidede genetiske studier til foreldre.
- Wilcox WR, Coulter CP, Schmitz ML (2015) Medfødte lem mangel lidelser. Clin Perinatol 42: 281-300.
- Bermejo SE, Cuevas L, Amar E (2011) Amelia: en multisenter deskriptiv epidemiologisk studie i et stort datasett fra International Clearinghouse For Birth Defects Surveillance And Research, og oversikt over litteraturen. Er J Med Genet C Semin Med Genet 157: 288-304.
- Zlotogora JSM, Shabany YO, Jarallah RY (1993) Syndrom av tetraamelia med lungehypoplasi. Er J Med Genet 47: 570-571.
- Zimmer EZ (1985) Tetra-amelia med flere misdannelser hos seks mannlige fostre i en slekt. Europ. J Pediat 144: 412-414.
- Rosenak D (1991) Tilbakevendende tetraamelia og pulmonal hypoplasi med flere misdannelser hos sibs. Er J Med Genet 38: 25-28.
- Gershoni BR (1990) Roberts syndrom eller «X-bundet amelia»? . Er J Med Genet 37: 569-572.
- Kosaki K, Jones MC, Stayboldt C (1996) Zimmer phocomelia: avgrensning ved hovedkoordinatanalyse. Er J Med Genet 66: 55-59.
- Niemann S(2004) Homozygot wnt3 mutasjon forårsaker tetra-amelia i en stor consanguineous familie. Er J Hum Genet 74: 558-563.
- Krahn M (2005) Tetra-amelia og lunge aplasi syndrom: rapport av en ny familie og utelukkelse av kandidatgener. Clin Genet 68: 558-560.
- Szenker-RE, Altunoglu U (2018) rspo2-hemming av RNF43 OG ZNRF3 styrer utvikling av lemmer uavhengig av LGR4 / 5 / 6. Natur 557: 564-569.