Periodiske Lammelser
en klinisk nyttig klassifisering av primære periodiske lammelser, vist i Tabell 1, inkluderer hypokalemiske, hyperkalemiske og paramyotoniske former.
Tabell 1. Primær Periodisk Lammelse (modifisert Fra Jurkat-Rott og Lehmann-Horn) (Åpent Bord i et nytt vindu)
|
Sykdom |
Gene |
Protein |
Arv |
Mutasjon |
|
HyperPP |
SCN4A |
Nav1.4 |
Dominant |
Fin |
|
NormoPP |
Fin (ω-pore) |
|||
|
Paramyotoniacongenita |
Fin |
|||
|
HypoPP Type II |
Fin (ω-pore) |
|||
|
HypoPP Bestill |
CACNA1S |
Cav1.1 |
Dominant |
Gevinst(ω-pore) |
|
ThyrotoxicPP |
KCNJ18 |
Kir2.18 |
Dominant |
Tap |
|
Andersen-Tawil syndrom |
KCNJ2 |
Kir2.1 |
Dominant |
Tap |
det fysiologiske grunnlaget for slap svakhet er inexcitability av muskelmembranen (dvs.sarcolemma). Endring av serumkaliumnivå er ikke hoveddefekten i PRIMÆR PP; den endrede kaliummetabolismen er et resultat av PP. I primær og tyrotoksisk PP oppstår slap lammelse med relativt små endringer i serumkaliumnivået, mens i sekundær PP er serumkaliumnivået markert unormalt.
Ingen enkelt mekanisme er ansvarlig for denne gruppen av lidelser. Dermed er de heterogene, men deler noen fellestrekk. Svakheten er vanligvis generalisert, men kan være lokalisert. Kranialmuskulatur og respiratoriske muskler blir vanligvis spart. Strekkreflekser er enten fraværende eller redusert under angrepene. Muskelfibrene er elektrisk uutslettelige under angrepene. Muskelstyrke er normalt mellom anfallene, men etter noen år utvikles en viss grad av fast svakhet i VISSE TYPER PP (spesielt primær PP). Alle former for PRIMÆR PP (unntatt Becker myotonia congenita ) er enten autosomal dominant arvelig eller sporadisk (mest sannsynlig som følge av punktmutasjoner).
Spenningsfølsomme ionekanaler regulerer tett generering av handlingspotensialer (korte og reversible endringer av spenningen til cellemembraner). Disse er selektivt og variabelt permeable ionekanaler. Energiavhengige ionetransportører opprettholder konsentrasjonsgradienter. Under genereringen av handlingspotensialer beveger natriumioner seg over membranen gjennom spenningsstyrte ionkanaler. Den hvilende muskelfibermembranen polariseres primært ved bevegelse av klorid gjennom kloridkanaler og repolariseres ved bevegelse av kalium. Natrium -, klorid-og kalsiumkanalopatier, som en gruppe, er forbundet med myotoni og PP. De funksjonelle underenhetene av natrium -, kalsium-og kaliumkanaler er homologe. Natriumkanalopatier forstås bedre enn kalsium-eller kloridkanalopatier. Alle former for FAMILIÆR PP viser den endelige mekanistiske vei som involverer avvikende depolarisering, inaktivering av natriumkanaler, og muskel fiber inexcitability.
Diskusjon i denne artikkelen omhandler primært natrium -, kalsium-og kaliumkanalopatier samt sekundære FORMER FOR PP. Kloridkanalopatier er ikke forbundet med episodisk svakhet og diskuteres mer detaljert i artiklene om myotoniske lidelser.
Oppsummering av kanal dysfunksjon i ulike TYPER PP
Med HyperPP rask kanal inaktivering, mutasjoner er vanligvis plassert i de indre delene av transmembrane segmenter eller i de intracellulære sløyfer påvirker dokkingssteder for rask inaktiverende partikkel, og dermed svekke rask kanal inaktivering fører til vedvarende Na + strøm.
med HypoPP hyperpolariseringsaktivert kationlekkasje som motvirker K+-likerettende strøm, forårsaker mutasjoner den ytterste arginin – eller lysinsubstitusjon.
Med NormoPP depolarisering-aktivert kationlekkasje er mutasjoner i dypere steder av spenningssensor av domene II ved kodon R675.
Ionekanal dysfunksjon er vanligvis godt kompensert med normal eksitasjon, og flere utløsere er ofte nødvendige for å produsere muskel inexcitability på grunn av vedvarende membran depolarisering.
Glukose og kaliuminntak har motsatte effekter i disse lidelsene. I HyperPP utløser kaliuminntaket angrepet, mens glukose forbedrer det. I kontrast fremkaller glukose hypokalemiske angrep og kalium er behandlingen for angrepet.
Merk bildet nedenfor.
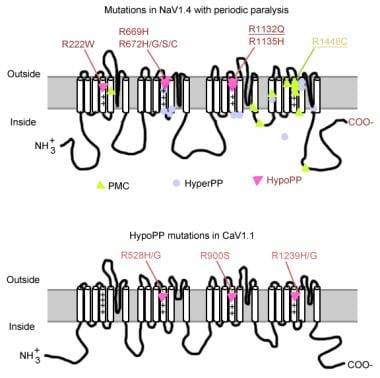 Mutasjoner i periodisk lammelse.
Mutasjoner i periodisk lammelse. muskelnatriumkanalgen
natriumkanalen har en alfa-underenhet og en beta-underenhet. Alfa-underenheten til natriumkanalen er et 260-kd glykoprotein bestående av ca. 1800-2000 aminosyrer. Denne kanalen er sterkt bevart evolusjonært fra Drosophila til menneske. Den har 4 homologe domener (I-IV) som bretter seg for å danne en sentral pore, hver med 225-325 aminosyrer. Hvert domene består av 6 hydrofobe segmenter (S1-S6) som krysser cellemembranen. Hovedfunksjonene til kanalen inkluderer spenningsfølsom gating, inaktivering og ion selektivitet. Den ekstracellulære sløyfen mellom S5 Og S6 faller inn i plasmamembranen og deltar i dannelsen av porene. S4-segmentet inneholder positivt ladede aminosyrer i hver tredje posisjon og fungerer som en spenningssensor. Konformasjonsendringer kan forekomme under depolarisering, noe som resulterer i aktivering og inaktivering av kanalen. Den cellulære sløyfen MELLOM domene III-S6 OG domene IV-S1 fungerer som en inaktiverende gate.
natriumkanalen har 2 porter (aktivering og inaktivering) og kan eksistere i 3 tilstander. I hvile med membranen polarisert, er aktiveringsporten lukket og inaktiveringsporten åpnes. Ved depolarisering åpnes aktiveringsporten, slik at natriumioner kan passere gjennom ionkanalen og også utsette et dockingssted for inaktiveringsporten. Ved fortsatt depolarisering lukkes inaktiveringsporten, blokkerer innføringen av natrium i cellen og forårsaker at kanalen går inn i hurtiginaktiveringstilstanden. Denne inaktiveringen av kanalen gjør at membranen blir repolarisert, noe som resulterer i en retur til hvilestatus med aktiveringsporten lukket og inaktiveringsporten åpnet. To inaktiveringsprosesser forekommer i pattedyrskjelettmuskulatur: Rask inaktivering innebærer å avslutte handlingspotensialet og virker på en millisekund tidsskala. Langsom inaktivering tar sekunder til minutter og kan regulere befolkningen av spennende natriumkanaler.
natriumkanalmutasjoner som forstyrrer rask og langsom inaktivering, er vanligvis forbundet med en fenotype Av HyperPP og myotoni, hvor mutasjoner som forbedrer langsom eller rask inaktivering forårsaker tap av natriumkanalfunksjon, forårsaker HypoPP.
Mutasjoner av natriumkanalgenet (SCN4A) har flere generelle egenskaper. De fleste mutasjonene er i» inaktiverende «linker mellom repeter III OG IV, i «spenningsfølende» segment S4 av repeat IV eller ved den indre membranen hvor de kan svekke dockingsstedet for inaktiveringsporten. Den kliniske fenotypen varierer ved spesifikk aminosyresubstitusjon, og mens noe overlapping kan forekomme mellom hyperkalemisk PP, paramyotonia congenita (PC) og kaliumforverrede myotonier( PAM), er de 3 fenotypene generelt distinkte (som beskrevet nedenfor). Nesten alle mutantkanaler har nedsatt hurtiginaktivering av natriumstrøm. De fleste pasienter er følsomme for systemisk kalium eller kald temperatur.
To populasjoner av kanaler eksisterer, mutant og villtype; nedsatt hurtiginaktivering resulterer i langvarig depolarisering av mutantmuskelfibermembraner og kan forklare de 2 kardinale symptomene på disse lidelsene, myotoni og svakhet. I hyperkalemisk PP oppstår en funksjonsforsterkning i mutantkanalregulering, noe som resulterer i økt natriumstrøm for mye depolarisering av den berørte muskelen. Mild depolarisering (5-10 mV) av myofibermembranen, som kan være forårsaket av økte ekstracellulære kaliumkonsentrasjoner, resulterer i at mutantkanalene opprettholdes i ikke-aktivert modus. Den vedvarende innadgående natriumstrømmen forårsaker repeterende avfyring av villtype natriumkanaler, som oppfattes som stivhet (dvs.myotoni).
hvis en mer alvorlig depolarisering (20-30 mV) er tilstede, er både normale og unormale kanaler festet i en tilstand av inaktivering, noe som forårsaker svakhet eller lammelse. Dermed kan subtile forskjeller i alvorlighetsgraden av membrandepolarisering gjøre forskjellen mellom myotoni og lammelse. Temperaturfølsomhet er et kjennetegn PÅ PC. Kald forverrer myotoni og induserer svakhet. En rekke mutasjoner er forbundet med denne tilstanden, 3 av dem på samme sted (1448) I s4-segmentet. Disse mutasjonene erstatter arginin med andre aminosyrer og nøytraliserer denne høyt konserverte s4 positive ladningen. Mutasjoner av disse rester er den vanligste årsaken TIL PC. Noen av de mulige mekanismene som er ansvarlige for temperaturfølsomhet inkluderer følgende:
-
Temperaturen kan differensielt påvirke konformasjonsendringen i mutantkanalen.
-
Lavere temperaturer kan stabilisere mutantkanalene i unormal tilstand.
-
Mutasjoner kan endre følsomheten til kanalen til andre cellulære prosesser, som fosforylering eller andre budbringere.
de fleste tilfeller av hyperkalemisk PP skyldes 2 mutasjoner I SCN4A, T704M OG M1592V. Mutasjoner i natriumkanalen, spesielt ved rester 1448 og 1313, er ansvarlige for paramyotonia congenita. En liten andel av hypokalemiske periodiske lammelsestilfeller er assosiert med mutasjoner ved kodon 669 og 672 (HypoPP2). I HypoPP2 øker natriumkanalmutasjoner inaktivering for å gi et netto tap av funksjonsdefekt.
Normokalemic PP ligner Både HyperPP (kalium følsomhet) Og HypoPP (varighet av angrep) og er forårsaket AV SCN4A mutasjoner på en dypere plassering av spenningssensor DII ved kodon 675. R675-mutasjoner skiller Seg fra HypoPP ved at disse mutasjonene resulterer i depolariseringsaktivert poregenerering av ω-strøm med reversert spenningsavhengighet da dette stedet eksponeres for ekstracellulære steder ved sterkere depolarisering.
Kalsiumkanalgen
kalsiumkanalgenet (CACNL1A3) er et kompleks av 5 underenheter (alfa-1, alfa-2, beta, gamma og delta). Dihydropyridin (DHP) – reseptoren for skjelettmuskulatur er hovedsakelig lokalisert i den tverrgående rørformede membranen. Alfa-1-underenheten har bindingssteder FOR DHP-legemidler og utfører langsom l-type kalsiumstrøm. Det deltar også i eksitasjons-sammentrekning (EC) kobling og fungerer som en spenningssensor gjennom sin kobling med ryanodinreseptoren av sarkoplasmatisk retikulum (dvs.kalsiumutløsningskanal). Eventuelle endringer i membranpotensialet er knyttet til intracellulær kalsiumfrigivelse, noe SOM muliggjør ec-kobling. Punktmutasjoner I DHP-reseptor / kalsiumkanal alfa-1-underenhet forårsaker hypokalemisk PP (HypoPP1). To mutasjoner AV cacna1s-genet, R528H OG R1239H, er ansvarlige for de fleste tilfeller av hypokalemisk PP.
den fysiologiske basis av sykdommen er fortsatt ikke forstått, men er mer sannsynlig på grunn av en svikt i eksitasjon snarere enn en svikt I EC kopling. Hypokalemiindusert depolarisering kan imidlertid redusere kalsiumutslipp og påvirke spenningskontrollen av kanalen direkte eller indirekte gjennom inaktivering av natriumkanalen. Insulin og adrenalin kan virke på en lignende måte. Mutasjoner av kalsiumkanalgenet har noen likheter MED SCN4A-mutasjoner. Mutasjoner modifiserer kanalinaktivering, men ikke spenningsavhengig aktivering. Opptak fra myotube kulturer fra berørte pasienter viste en 30% reduksjon I DHP-sensitiv l-type kalsiumstrøm. Kanaler er inaktivert ved lave membranpotensialer.
Kalsiumkanalmutasjoner forårsaker tap av funksjon manifestert som redusert strømtetthet og langsommere inaktivering. Hvordan denne inaktiveringen er relatert til hypokalemi-induserte angrep, er ikke forstått. I DET minste i r528h-mutasjon oppstår en mulig sekundær kanalopati, knyttet til en reduksjon I ATP-sensitiv kaliumstrøm fra endret kalsiumhomeostase. De lavere strømningene forbundet MED cacnl1a3-mutasjoner kan lett endre intracellulær kalsiumhomeostase, noe som kan påvirke egenskapene Og ekspresjonen Av k+-kanaler, spesielt KATP (ATP-sensitiv kaliumkanal) som tilhører innad likeretterklasse av kanaler. Insulin virker Også I HypoPP ved å redusere denne innad likeretter k + strøm.
Spenningsfølerladningstap står for De fleste Tilfeller Av HypoPP. Natrium – og kalsiumkanaler har homologe poredannende alfa-underenheter. Punktmutasjoner I CACNL1A3 og SCN4A påvirker argentinske rester I s4 spenningssensorene til disse kanalene. Arginin mutasjoner I s4 segmenter er ansvarlig for 90% Av HypoPP tilfeller.
Spenningsfølerladningstap står for De fleste Tilfeller Av HypoPP. Natrium – og kalsiumkanaler har homologe poredannende underenheter. Nesten alle mutasjonene I Cav1.1 (HypoPP-1) Og Nav1.4 (HypoPP-2) nøytraliserer en positivt ladet aminosyre i en av de ytterste argininer eller lysiner av spenningssensorer. Nav1.4 mutasjoner er oftest plassert i spenningssensorene I I, II OG III gjentakelser, forårsaker en kationlekkasje.
Substitusjon av ytterste arginin med en mindre aminosyre som glysin åpner en ledende vei ved hyperpolarisert potensial, noe som resulterer i en innadgående kationstrøm (kationlekkasje eller ω strøm for å skille fra (ω-) gjennom ioneledende pore, er en hyperpolariserings–aktivert strøm av monovalente kationer gjennom S4-gatepore som motvirker likerettende k+ strømmer) depolarisering eller destabilisering av hvilepotensialet.
S4-segmentet beveger seg utover under depolarisering som lukker den ledende banen. Muskelfibre med alvorlige spenningssensormutasjoner depolariseres ikke bare under hypokalemi, men også på kaliumnivåer i det normale området, og forklarer interiktal og permanent svakhet. Alvorlig myopati med fettutskifting av muskelvev er ofte funnet hos pasienter Med Cav1. 1 R1239H (DIV-mutasjoner).
Glukokortikosteroider forårsaker HypoPP ved å stimulere Na + K + ATPase mediert av insulin og amylin.
Kaliumkanalgen
Innadretting er en viktig egenskap For Kir-kanaler. Utbedring innebærer spenningsavhengig ledning-poreblokkering av pore med polyaminer og Mg++ under depolarisering, og denne blokkeringen fjernes under potensiell gradient under hyperpolarisering. Kaliumkanalmutasjoner er sett I Andersen-Tawil syndrom og tyrotoksisk PP.
Triaden av dysmorfe egenskaper, periodisk lammelse og hjertearytmier karakteriserer Andersen-Tawil syndrom. Dette syndromet er forbundet med mutasjoner I kcnj2-genet. Kcnj2-genet koder For Den innadrettede kaliumkanalen Kir2. 1. Kaliumkanalmutasjoner I KCNE3 er rapportert å forårsake hypokalemisk PP, men dette er ikke sannsynliggjort.
Mutasjoner I Kir2. 6 forårsaker følsomhet for tyrotoksisk PP. Episodisk svakhet sett i thyrotoxic PP er lik Den sett I HypoPP og Andersen-Tawil syndrom. Denne lidelsen er mest utbredt I Asiater og latinamerikanske menn. Thyrotoxic PP er en genetisk lidelse unmasked av thyrotoxicosis. Kir2.6 uttrykkes primært i skjelettmuskulatur. Triiodothyronin forbedrer kcnj18 transkripsjon, noe som kan drive forbedret uttrykk For Kir2. 6. PKC aktiveres under tyrotoksikose på grunn av økt PIP2-omsetning og Kir-kanaler interagerer direkte med PIP2 under normal gating. I Andersen-Tawil syndrom er det redusert PIP2 affinitet. I tyrotoksisk PP endrer Ingen av mutasjonene Kir2. 6-utbedring.