Gynaecology & Obstetrics Case report
trefwoorden
Tetra-amelia; Malformation; Genotype; fenotype.
Inleiding
afwijkingen van de ledematen vormen een belangrijke groep congenitale pathologieën die gekarakteriseerd worden door hypoplasie of volledige aplasie van één of meer botten van de ledematen. Bij ongeveer 1 op de 1.300 tot 2.000 geboorten treden allerlei afwijkingen van de ledematen op. Deze afwijkingen van de ledematen kunnen worden geïsoleerd of geassocieerd met andere afwijkingen . Tetraamelia syndroom is zeldzaam en grijze gebieden blijven.
we melden twee gevallen van tetra-amelia in een niveau II Moederschap in Dakar (Senegal) die vergelijkbaar zijn met tetraamelia-1 (chromosoom 17q21), tetraamelia-2 (chromosoom 8q23) en Robert ‘ s syndroom (chromosoom 8p21). Dit illustreert de moeilijkheid in het correleren van fenotype en betrokken genen.
Case Reports
Case 1
MS. AD was een 44-jarige moeder verwezen naar onze afdeling op 36 weken zwangerschap met ernstige pre-eclampsie en foetale afwijkingen. Ze was vijf para ‘ s zonder voorgeschiedenis van foetale afwijkingen. Ze rookte nu niet en had nooit gerookt noch alcohol gedronken. Ze was niet blootgesteld aan passief roken. Ze was in een derdegraads bloedverwantschap voor al haar kinderen. Ms. AD had negatief getest op hepatitis B, HIV en syfilis. Ze was beschermd tegen het rubellavirus en was niet eerder blootgesteld aan Toxoplasma gondii. Echografie die laat na 33 weken en 35 weken van de zwangerschap werd uitgevoerd, vond oligoamniose en hydrocefalie, evenals agenese van de ledematen. Voorschriften tijdens de zwangerschap omvatten de toediening van ijzer en foliumzuur, evenals de toediening van sulfadoxine pyrimethamine. De laatste werd voorgeschreven op 18 weken en vervolgens 26 weken als onderdeel van het anti-malaria profylaxe beleid voor zwangere vrouwen. De symphyseal-fundal hoogte gemeten 28 cm. Vanwege ernstige kenmerken van pre-eclampsie; ze werd onmiddellijk in het ziekenhuis opgenomen en geobserveerd in een weeën-en afleveringseenheid. Daarna kreeg ze in eerste instantie IV magnesiumsulfaat om eclampsie en antihypertensiva te voorkomen om de systolische bloeddruk onder 160 mmHg en de diastolische bloeddruk onder 105 mmHg te houden.
het besluit tot onmiddellijke levering van een keizersnede werd genomen. Een levend geboren 2,150 gram werd geëxtraheerd die vervolgens binnen 10 minuten stierf. Voor de dood vertoonde het lichaam kruipende bewegingen. Verschillende uitwendige afwijkingen werden geïdentificeerd (figuur 1), waaronder volledige agenese van alle vier de ledematen, hydrocephalus met een hoofdomtrek van 39 cm. Op het gezicht was er hypertelorisme met een coloboom van de oogleden, milde exophthalmos en aniridië. De mond was als een omgekeerde V zonder duidelijke afbakening van de lippen en de neus was rudimentair. De pasgeborene was verstoken van integumenten (haar en wenkbrauwen). De oren werden gereduceerd tot schetsen die eruit zagen als spleten. De nek was kort. De stam werd gereduceerd tot een 26 cm lange conische structuur met een navelstreng aan de onderkant. De borst was smal. Net onder de navel, was de stam naar achteren verwijd aanwezig op de vloer van de caudale paal, een middellijn met recessies en een ontluikende die kan overeenkomen met een fallus van onbepaald type. Agenese van het bekken, genitaliën en anale imperforatie werden opgemerkt. Foetale pathologie is niet uitgevoerd. Echter, overlijden binnen 10 minuten na de bevalling en het kegelvormige uiterlijk van de borst kan wijzen op longafwijkingen.
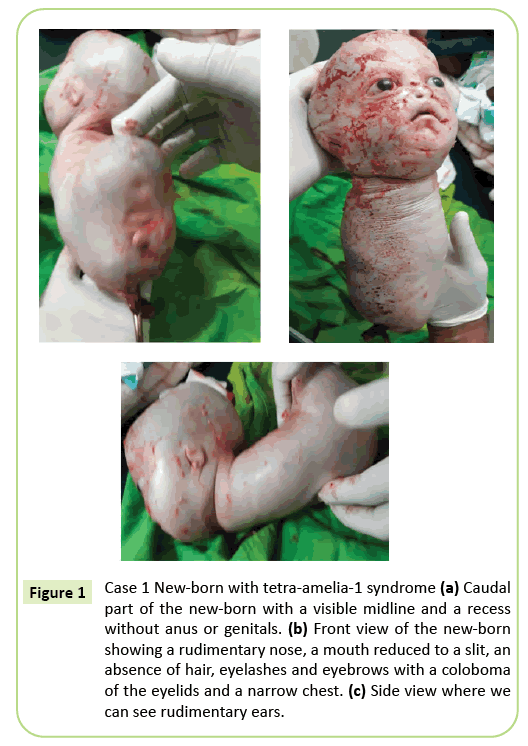
figuur 1: Geval 1 pasgeborene met tetra-amelia-1 syndroom (a) caudaal deel van de pasgeborene met een zichtbare middenlijn en een uitsparing zonder anus of genitaliën. B) vooraanzicht van de pasgeborene met een rudimentaire neus, een mond gereduceerd tot een spleet, een afwezigheid van haar, wimpers en wenkbrauwen met een coloboom van de oogleden en een smalle borst. (C) zijaanzicht waar we rudimentaire oren kunnen zien.
zaak 2
het tweede geval was een 22-jarige primigravida die naar onze faciliteit werd verwezen voor een ultrasone scan na 37 weken zwangerschap. Ze had geen bloedverwantschap. Ze had negatief getest op hepatitis B, HIV en syfilis. Ze is niet getest op toxoplasmose en rodehond. Er is geen echografie gedaan tijdens haar zwangerschap. Het klinische onderzoek was consistent met foetale groeivertraging (Fundal Lengte: 26 cm). Ultrasone bevindingen toonden aan dat het opperarmbeen vervormd was met een lengte van 23,9 mm, wat overeenkomt met 17 weken zwangerschap. Er was agenese van het dijbeen. De iliacale vleugels waren zichtbaar op echografie. Er werden geen long-of hartafwijkingen vastgesteld. De levering werd gestart. De pasgeborene had een vrouwelijk fenotype met een Apgar score van 9 op de 5e minuut. De morfologie van het hoofd en de stam was zonder bijzonderheid. De bovenste ledematen werden gereduceerd tot twee 3 cm lange stompen. Volledige agenese van de 2 onderste ledematen werd opgemerkt. Het was een symmetrische anomalie (Figuur 2).
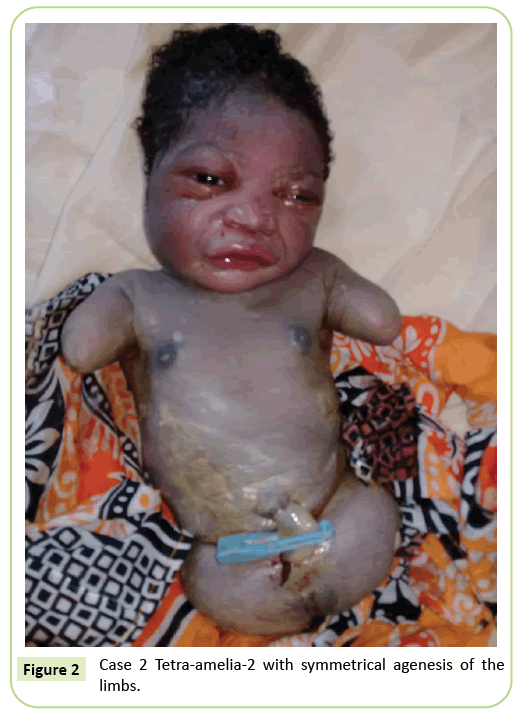
Figuur 2: geval 2 Tetra-amelia-2 met symmetrische agenese van de ledematen.
discussie
Bermejo-Sanchez et al. beschreven in 2011 de epidemiologie van congenitale amelia met behulp van gegevens verzameld uit 20 congenitale anomalieën surveillance programma ‘ s, uit alle continenten behalve Afrika, tussen 1968 en 2006. In totaal werden 326 gevallen van amelia geïdentificeerd onder 23.110.591 levendgeborenen, doodgeborenen en abortussen. De prevalentie was 1,41 / 100.000 .
Tetra-amelia verwijst naar de volledige afwezigheid van de ledematen en komt zeldzamer voor. Voor zover wij weten, wordt tetra-amelia-1 beschreven in 7 families. Het lijkt een autosomaal recessieve overerving te volgen. In alle families werd tetra-amelia-1 geassocieerd met ernstige misvormingen van de andere delen van het lichaam, waaronder het gezicht en het hoofd, anomalieën van het zenuwstelsel, skelet en genitaliën. De longen waren in veel gevallen onderontwikkeld, wat ademhaling moeilijk of onmogelijk maakt . Zimmer et al. gemeld in 1985 een sterk ingeteelde familie waarin 6 zuigelingen tetra-amelia-1 en hydrocefalie hadden. Ze beschreven in een van de foetussen een totale afwezigheid van bekkenbeen, gespleten lip en gehemelte, arrhinia en aplasie van de oren. Een bilaterale linkerlong, een persistent arterieel kanaal, een anale imperforatie werden ook opgemerkt. Foetale testen elimineerden de diagnose van Robert ‘ s syndroom . Andere gevallen uit de literatuur zijn onder andere die van Kosaki et al., in 1996, met een foetus van karyotype 46, XX met tetraphocomelia en ernstige pulmonale hypoplasie naast gezicht en hoofd anomalieën . Rosenak et al. beschreven een geval van tetra-amelia met ernstige pulmonale hypoplasie in twee foetussen van een niet-bloedverwantschap paar. Foetale testen sluiten diagnose van Robert ‘ s syndroom uit . Twee bijkomende gevallen werden gemeld door Zlotogora et al. in 1993. Beide patiënten stierven kort na de geboorte en de auteurs suggereerden het bestaan van pulmonale hypoplasie. Niemann et al. rapporteerde een bloedverwantschap Turkse familie waarin 4 van de 8 broers leden aan tetra-amelia. Naast de afwezigheid van de 4 ledematen, brachten de foetale onderzoeken van 3 foetussen meerdere anomalieën aan het licht: gespleten lippen en /of palatine, laparoschisis, pulmonale anomalieën, hypoplasie van het bekken, atresie van de choana ‘ s, vagina en anale imperforatie . Tot slot, in 2005, Krahn et al. beschreven 2 broers geboren uit inteelt ouders die lijden aan tetraamelia en ernstige pulmonale hypoplasie. De sleutelbenen en schouderbladen waren normaal bij de tweede foetus. Het karyotype was normaal .
Tetra-amelia-1-syndroom of TETAMS1 wordt veroorzaakt door een homozygote mutatie in het wnt3-gen op chromosoom 17q21 met een autosomaal recessieve overerving. Tetraamelia-2 syndroom (TETAMS2) wordt gekenmerkt door rudimentaire ledematen of een volledige afwezigheid van de ledematen, over het algemeen symmetrische en bilaterale agenese van de longen in sommige gevallen. Zijn ook gebruikelijke afwijkingen van het pulmonale vasculaire systeem en dysmorfieën, waaronder bilaterale gespleten lip en gehemelte, ankyloglossie, mandibulaire hypoplasie, microretrognathie en labioscrotal aplasie .
Szenker-Ravi, die 4 families van tetra-amelia met agenese of pulmonale hypoplasie bestudeerde, merkte een fenotypische heterogeniteit op met afwijkingen van de ledematen van verschillende ernst . Het exome rangschikken in deze 4 families heeft het mogelijk gemaakt om truncating homozygote veranderingen in het gen rspo2 te identificeren . Tetraamelia-2 syndroom wordt veroorzaakt door een homozygote mutatie in het rspo2 gen (610575) op chromosoom 8q23 .
het fenotype van het eerste geval dat in dit artikel wordt beschreven, komt overeen met een tetra-amelia-1-syndroom dat met name te wijten is aan de aanwezigheid van Hydrocefalie, anomalieën van de genitaliën en een rudimentaire neus. De smalle borst en vroege dood voor de 10e minuut van het leven wijzen op ernstige pulmonale hypoplasie. Dit geval benadrukt de fenotypische heterogeniteit met een ooglid coloboom, hypertelorisme, exoftalmos en zeldzame aanhangsels.
wij beschouwen het tweede geval in onze studie als een tetra-2-syndroom, gezien de symmetrische tetra-amelia met de aanwezigheid van stompen van de bovenste ledematen. De diagnose tetra-amelia moet vroeg worden gesteld tijdens de echografie. Daarom moet men zich bewust worden van het belang van echografie en het gebruik van 3D/4D om de screeningresultaten te verbeteren. De diagnose van een bekken massa op echografie in combinatie met amelia moet vermoeden voor spleno-gonadale fusie ledemaat defectsyndroom.
bovendien moeten het foetaal onderzoek en het foetaal testen met behulp van de evoluerende technologieën van chromosomale microarray en exoom-en genoomsequencing in onze settings worden aangemoedigd. Een betere karakterisering van de gevallen maakt het mogelijk om advies te geven aan koppels en een betere kennis van deze klinische anomalieën.
conclusie
Tetra-amelia syndroom is schaars en grijze gebieden blijven bestaan. Deze twee gevallen, vergeleken met wat reeds in de literatuur is beschreven, illustreren de fenotypische heterogeniteit van tetraamelia. Gezien de zeldzame incidentie van deze anomalieën, zou het belangrijk zijn om een internationaal register van anomalieën te creëren om gevallen te rapporteren en een steekproefbank op te zetten voor uitgebreide genetische studies aan ouders.
- Wilcox WR, Coulter CP, Schmitz ML (2015) Congenital limb deficiency disorders. Clin Perinatol 42: 281-300.
- Bermejo SE, Cuevas L, Amar E (2011) Amelia: A multi-center descriptive epidemiologic study in a large dataset from the International Clearinghouse for Birth defecten Surveillance and Research, and overview of the literature. Am J Med Genet C Semin Med Genet 157: 288-304.Zlotogora JSM, Shabany YO, Jarallah RY (1993) syndroom van tetraamelia met pulmonale hypoplasie. Am J Med Genet 47: 570-571.
- Zimmer EZ (1985) Tetra-amelia met meerdere misvormingen bij zes mannelijke foetussen in één verwant. Europ. J Kindergeneeskunde 144: 412-414.Rosenak D (1991) recidiverende tetraamelia en pulmonale hypoplasie met meerdere malformaties in SIB ‘ s. Am J Med Genet 38: 25-28.Gershoni BR (1990) Roberts syndroom of “X-linked amelia” ? . Am J Med Genet 37: 569-572.
- Kosaki K, Jones MC, Stayboldt C (1996) Zimmer phocomelia: delineation by principal coordinate analysis. Am J Med Genet 66: 55-59.Niemann S(2004) homozygote wnt3-mutatie veroorzaakt tetra-amelia in een grote verwantschapsfamilie. Am J Hum Genet 74: 558-563.Krahn m (2005)Tetra-amelia and lung aplasia syndrome: report of a new family and exclusion of candidate genes. Clin Genet 68: 558-560.
- Szenker-RE, Altunoglu U (2018) rspo2 remming van RNF43 en ZNRF3 regelt de ontwikkeling van de ledematen onafhankelijk van LGR4/5/6. Natuur 557: 564-569.