periodieke Paralyses
een klinisch bruikbare classificatie van primaire periodieke paralyses, weergegeven in Tabel 1, omvat hypokalemische, hyperkalemische en paramyotonische vormen.
Tabel 1. Primaire Periodieke Verlammingen (aangepast van Jurkat-Rott en Lehmann-Hoorn ) (Tabel Openen in een nieuw venster)
|
Ziekte |
Gen |
Eiwit |
Erfenis |
Mutatie |
|
HyperPP |
SCN4A |
Nav1.4 |
Dominante |
Fijn |
|
NormoPP |
Fijne (ω-porie) |
|||
|
Paramyotoniacongenita |
Fijn |
|||
|
HypoPP Type II |
Fijne (ω-porie) |
|||
|
HypoPP OM |
CACNA1S |
Cav1.1 |
Dominante |
Winst (ω-porie) |
|
ThyrotoxicPP |
KCNJ18 |
Kir2.18 |
Dominante |
Verlies |
|
Andersen-tawil beluistert syndroom |
KCNJ2 |
Kir2.1 |
Dominante |
Verlies |
De fysiologische basis van slappe zwakte is inexcitability van de spier membraan (dat wil zeggen, sarcolemma). Verandering van het serumkalium is niet het belangrijkste defect in primair PP; het veranderde kaliummetabolisme is een gevolg van PP. In primaire en thyrotoxische PP treedt slappe verlamming op met relatief kleine veranderingen in het serumkaliumniveau, terwijl in secundaire PP de serumkaliumniveaus duidelijk abnormaal zijn.
geen enkel mechanisme is verantwoordelijk voor deze groep van aandoeningen. Ze zijn dus heterogeen, maar hebben een aantal gemeenschappelijke kenmerken. De zwakte is meestal gegeneraliseerd, maar kan worden gelokaliseerd. Schedelspieren en ademhalingsspieren worden meestal gespaard. Stretch reflexen zijn afwezig of verminderd tijdens de aanvallen. De spiervezels zijn elektrisch onverwoestbaar tijdens de aanvallen. Spierkracht is normaal tussen aanvallen, maar na een paar jaar, een zekere mate van vaste zwakte ontwikkelt zich in bepaalde soorten PP (vooral primaire PP). Alle vormen van primaire PP (behalve Becker myotonia congenita) zijn ofwel autosomaal dominant geërfd of sporadisch (hoogstwaarschijnlijk voortkomend uit puntmutaties).
Spanningsgevoelige ionenkanalen regelen het genereren van actiepotentialen nauw (korte en omkeerbare veranderingen van de spanning van celmembranen). Dit zijn selectief en variabel doorlaatbare ionenkanalen. Energieafhankelijke ionentransporters behouden concentratiegradiënten. Tijdens de generatie van actiepotentialen, bewegen de natriumionen zich over het membraan door voltage-gated ionenkanalen. Het membraan van de rustspiervezel wordt hoofdzakelijk door de beweging van chloride door chloridekanalen gepolariseerd en door beweging van kalium repolarized. Natrium -, chloride-en calciumkanaalopathieën, als groep, worden geassocieerd met myotonie en PP. De functionele subeenheden van natrium -, calcium-en kaliumkanalen zijn homoloog. Natriumkanaalopathieën worden beter begrepen dan calcium-of chloridekanaalopathieën. Alle vormen van familiale PP tonen de definitieve mechanistische weg die afwijkende depolarisatie impliceren, het inactiveren van natriumkanalen, en spiervezel onvergeeflijkheid.
discussie in dit artikel behandelt voornamelijk de natrium -, calcium-en kaliumkanaalopathieën en secundaire vormen van PP. Chloride channelopathieën worden niet geassocieerd met episodische zwakte en worden in meer detail besproken in de artikelen over Myotone aandoeningen.
samenvatting van kanaaldisfunctie bij verschillende typen PP
met HyperPP snelle kanaalinactivatie bevinden mutaties zich gewoonlijk in de binnenste delen van transmembraansegmenten of in de intracellulaire lussen die de docking-plaatsen voor het snelle inactiverende deeltje beïnvloeden, waardoor snelle kanaalinactivatie wordt belemmerd, wat leidt tot aanhoudende Na+ – stroom.
bij HypoPP hyperpolarisatie-geactiveerde kationlek die K+ -rectificerende stroom tegenwerkt, veroorzaken mutaties buitenste arginine-of lysine-substitutie.
met normopp-depolarisatie-geactiveerd kationlek, bevinden mutaties zich op diepere locaties van de spanningssensor van domein II bij codon R675.
disfunctie van het ionenkanaal wordt gewoonlijk goed gecompenseerd door een normale excitatie, en bijkomende triggers zijn vaak nodig om spieronbekwaamheid te veroorzaken als gevolg van aanhoudende membraandepolarisatie.
Glucose-en kaliuminname heeft bij deze aandoeningen het tegenovergestelde effect. Bij HyperPP, veroorzaakt de inname van kalium de aanval, terwijl glucose het verbetert. Glucose daarentegen veroorzaakt hypokalemische aanvallen en kalium is de behandeling voor de aanval.
let op de afbeelding hieronder.
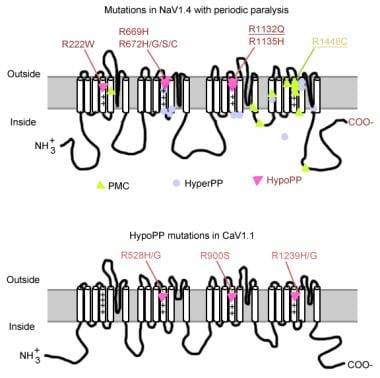 mutaties bij periodieke paralyse.
mutaties bij periodieke paralyse. gen voor het spiernatriumkanaal
het natriumkanaal heeft een alfa-en een bèta-subeenheid. De alpha-subeenheid van het natriumkanaal is een 260-KD glycoproteïne bestaande uit ongeveer 1800-2000 aminozuren. Dit kanaal wordt evolutionarily hoogst behouden van Drosophila aan mens. Het heeft 4 homologe domeinen (I-IV) die vouwen om een centrale porie te vormen, elk met 225-325 aminozuren. Elk domein bestaat uit 6 hydrophobic segmenten (S1-S6) die het celmembraan doorkruisen. De belangrijkste functies van het kanaal omvatten spanningsgevoelige gating, inactivatie, en ionenselectiviteit. De extracellulaire lijn tussen S5 en S6 duikt in het plasmamembraan en neemt deel aan de vorming van de porie. Het S4-segment bevat positief geladen aminozuren op elke derde positie en functioneert als spanningssensor. Veranderingen in de bouw kunnen optreden tijdens depolarisatie, resulterend in activering en inactivering van het kanaal. De cellulaire lijn tussen domein III-S6 en domein IV-S1 doet dienst als inactiverende poort.
het natriumkanaal heeft 2 poorten (activering en inactivering) en kan bestaan in 3 toestanden. In rust met het gepolariseerde membraan wordt de activeringspoort gesloten en de inactivatiepoort geopend. Met depolarisatie, opent de activeringspoort, die natriumionen toestaat om door het ionenkanaal te gaan en ook een docking plaats voor de inactivatiepoort blootstellen. Bij voortdurende depolarisatie sluit de inactivatiepoort, waardoor de ingang van natrium in de cel wordt geblokkeerd en het kanaal de fast-inactivatietoestand ingaat. Deze inactivatie van het kanaal staat het membraan toe om repolarized te worden, resulterend in een terugkeer naar de rustende staat met de gesloten activeringspoort en de geopende inactivatiepoort. Twee inactivatieprocessen komen voor in skeletspieren van zoogdieren: snelle inactivatie impliceert het beëindigen van het actiepotentieel en handelt op een milliseconde tijdschaal. Langzame inactivatie duurt seconden tot minuten en kan de populatie van prikkelbare natriumkanalen regelen.
natriumkanaalmutaties die de snelle en langzame inactivatie verstoren, worden gewoonlijk geassocieerd met een fenotype van HyperPP en myotonie, waarbij als mutaties die de langzame of snelle inactivatie versterken en leiden tot verlies van de natriumkanaalfunctie HypoPP veroorzaken.
mutaties van het natriumkanaalgen (SCN4A) hebben verschillende algemene kenmerken. De meeste mutaties bevinden zich in de “inactiverende” linker tussen herhalingen III en IV, in het “voltage-sensing” segment S4 van herhaal IV of bij het binnenste membraan waar zij de docking plaats voor de inactiveringspoort zouden kunnen aantasten. Het klinische fenotype verschilt door specifieke aminozuursubstitutie en hoewel er enige overlap kan optreden tussen hyperkalemische PP, paramyotonia congenita (PC) en kalium-verergerde myotonia (PAM), zijn de 3 fenotypen over het algemeen verschillend (zoals hieronder beschreven). Bijna alle mutantkanalen hebben de snelle inactivering van de natriumstroom verstoord. De meeste patiënten zijn gevoelig voor systemische kalium of voor koude temperatuur.
er bestaan twee populaties van kanalen, mutant en wild-type; de verminderde snelle inactivatie leidt tot langdurige depolarisatie van de gemuteerde spiervezelmembranen en kan de twee belangrijkste symptomen van deze aandoeningen, myotonie en zwakte, verklaren. In hyperkalemic PP, komt een aanwinst van functie in mutant kanaal gating voor, resulterend in een verhoogde natriumstroom bovenmatig depolariserend de beà nvloede spier. Milde depolarisatie (5-10 mV) van het myofibermembraan, die kan worden veroorzaakt door verhoogde extracellulaire kaliumconcentraties, resulteert in de mutantkanalen die in de niet-geïnactiveerde modus worden gehandhaafd. De aanhoudende inkomende natriumstroom veroorzaakt repetitieve verbranding van de wild-type natriumkanalen, die wordt waargenomen als stijfheid (dwz, myotonie).
als een ernstigere depolarisatie (20-30 mV) aanwezig is, worden zowel normale als abnormale kanalen gefixeerd in een staat van inactivering, wat zwakte of verlamming veroorzaakt. Aldus, kunnen de subtiele verschillen in strengheid van membraandepolarisatie het verschil tussen myotonia en verlamming maken. Temperatuurgevoeligheid is een kenmerk van PC. Verkoudheid verergert myotonie en veroorzaakt zwakte. Een aantal veranderingen worden geassocieerd met deze voorwaarde, 3 van hen op dezelfde plaats (1448) in het segment S4. Deze veranderingen vervangen arginine met andere aminozuren en neutraliseren deze hoogst behouden S4 positieve Last. Mutaties van deze residuen zijn de meest voorkomende oorzaak van PC. Enkele van de mogelijke mechanismen verantwoordelijk voor temperatuurgevoeligheid omvatten het volgende:
-
temperatuur kan de conformationele verandering in het mutantkanaal verschillend beïnvloeden.
-
lagere temperaturen kunnen de mutantkanalen stabiliseren in een abnormale toestand.
-
de veranderingen kunnen de gevoeligheid van het kanaal aan andere cellulaire processen, zoals phosphorylation of tweede boodschappers veranderen.
de meeste gevallen van hyperkalemic PP zijn toe te schrijven aan 2 veranderingen in SCN4A, T704M, en M1592V. Mutaties in het natriumkanaal, vooral bij residuen 1448 en 1313, zijn verantwoordelijk voor paramyotonia congenita. Een klein deel van hypokalemische periodieke paralysegevallen wordt geassocieerd met mutaties bij codons 669 en 672 (HypoPP2). In HypoPP2, verbeteren de veranderingen van het natriumkanaal inactivatie om een netto verlies van functiedefect te veroorzaken.
Normokalemische PP lijkt op zowel HyperPP (kaliumgevoeligheid) als HypoPP (duur van aanvallen) en wordt veroorzaakt door scn4a mutaties op een diepere locatie van spanningssensor DII bij codon 675. De veranderingen r675 verschillen van HypoPP in die zin dat deze veranderingen in depolarisatie resulteren-geactiveerde gating porie die ω-stroom met omgekeerde voltageafhankelijkheid produceren aangezien deze plaats aan extracellulaire plaatsen bij sterkere depolarisatie wordt blootgesteld.
Calciumkanaalgen
het calciumkanaalgen (CACNL1A3) is een complex van 5 subeenheden (alfa-1, alfa-2, bèta, gamma en delta). De dihydropyridine (DHP) receptor van de skeletspier bevindt zich voornamelijk in het transversale buisvormige membraan. De alpha – 1 subeenheid heeft bindingsplaatsen voor DHP drugs en voert de langzame L-type calciumstroom uit. Het neemt ook deel aan excitatie-contractie (EC) koppeling en fungeert als een spanningssensor door zijn koppeling met de ryanodine receptor van sarcoplasmatisch reticulum (dat wil zeggen, calcium release kanaal). Om het even welke veranderingen in het membraanpotentieel zijn verbonden met intracellular calciumversie, die EG-koppeling toelaten. Puntmutaties in DHP receptor/calciumkanaal alfa-1 subeenheid veroorzaken hypokalemische PP (HypoPP1). Twee mutaties van CACNA1S gen, R528H en R1239H, zijn verantwoordelijk voor de meeste gevallen van hypokalemische PP.
de fysiologische basis van de ziekte is nog steeds niet bekend, maar is eerder te wijten aan het falen van de excitatie dan aan het falen van de EG-koppeling. Hypokaliëmie-geïnduceerde depolarisatie kan echter de afgifte van calcium verminderen, waardoor de spanningsregeling van het kanaal direct of indirect wordt beïnvloed door inactivering van het natriumkanaal. Insuline en adrenaline kunnen op een vergelijkbare manier werken. De veranderingen van het calciumkanaalgen hebben sommige gelijkenissen met de mutaties van SCN4A. Mutaties wijzigen kanaalinactivatie maar niet spanningsafhankelijke activering. Opnames van myotube culturen van getroffen patiënten toonden een 30% vermindering van de DHP-gevoelige L-type calciumstroom. Kanalen worden geïnactiveerd bij een laag membraanpotentieel.
Calciumkanaalmutaties veroorzaken een functieverlies dat zich manifesteert als een verminderde stroomdichtheid en een tragere inactivatie. Hoe deze inactivatie gerelateerd is aan hypokaliëmie-geïnduceerde aanvallen is niet begrepen. Ten minste in r528h-mutatie, treedt een mogelijke secundaire kanaalopathie op, gebonden aan een vermindering van de ATP-gevoelige kaliumstroom van veranderde calciumhomeostase. De lagere stromen geassocieerd met CACNL1A3-mutaties kunnen de intracellulaire calciumhomeostase enigszins veranderen, wat de eigenschappen en expressie van K+ – kanalen kan beïnvloeden, in het bijzonder KATP (ATP-sensitive potassium channel) behorend tot de naar binnen gelijkrichter klasse van kanalen. Insuline werkt ook in HypoPP door het verminderen van deze naar binnen gelijkrichter K + stroom.
spanningsverlies van de sensor is verantwoordelijk voor de meeste gevallen van HypoPP. Natrium-en calciumkanalen hebben homologe porievormende alfa-subeenheden. Puntmutaties in CACNL1A3 en SCN4A beïnvloeden Argentijnse residuen in de S4 spanningssensoren van deze kanalen. Argininemutaties in S4-segmenten zijn verantwoordelijk voor 90% van de HypoPP-gevallen.
spanningsverlies van de sensor is verantwoordelijk voor de meeste gevallen van HypoPP. Natrium-en calciumkanalen hebben homologe porievormende α-subeenheden. Bijna alle mutaties in Cav1.1 (HypoPP-1) en Nav1.4 (HypoPP-2) neutraliseren een positief geladen aminozuur in een van de buitenste arginines of lysines van spanningssensoren. De Nav1.4 mutaties bevinden zich meestal in de spanningssensoren van I, II en III herhalingen, waardoor een kationlek ontstaat.
substitutie van buitenste arginine door een kleiner aminozuur zoals glycine opent een geleidende weg bij hyperpolarized potentieel, resulterend in een binnenwaarts kation stroom (kationlek of ω stroom om te onderscheiden van (ω-) Door iongeleidende porie, is een hyperpolarisatie–geactiveerde stroom van monovalente kationen door S4 gating porie tegen rectificerende K+ stromen) depolarisatie of destabilisatie van het rustpotentieel.
S4-segment beweegt naar buiten tijdens depolarisatie en sluit de geleidende weg. Spiervezels met ernstige voltagesensormutaties worden niet alleen tijdens hypokaliëmie gedepolariseerd, maar ook bij kaliumniveaus in het normale bereik, wat interictale en permanente zwakte verklaart. Ernstige myopathie met vetvervangende spierweefsel wordt vaak gevonden bij patiënten met Cav1.1 R1239H (DIV-mutaties).
glucocorticosteroïden veroorzaken HypoPP door het stimuleren van Na+ K+ ATPase gemedieerd door insuline en Amyline.
Kaliumkanaalgen
binnenwaartse rectificatie is een belangrijke eigenschap van Kir-kanalen. Rectificatie impliceert voltage-afhankelijke geleiding-porieblokkering van porie met polyamines en Mg++ tijdens depolarisatie, en deze blokkade wordt verwijderd tijdens potentiële gradiënt tijdens hyperpolarisatie. Kaliumkanaalmutaties worden gezien bij Andersen-Tawil syndroom en thyrotoxische PP.
de triade van dysmorfe kenmerken, periodieke paralyse en hartritmestoornissen karakteriseert het Andersen-Tawil-syndroom. Dit syndroom wordt geassocieerd met mutaties in het gen KCNJ2. Het kcnj2 gen codeert het naar binnen gerichte rectificerende kaliumkanaal Kir2. 1. Van kaliumkanaalmutaties in KCNE3 wordt gemeld dat ze hypokalemische PP veroorzaken, maar dit is niet onderbouwd.
mutaties in Kir2.6 veroorzaken gevoeligheid voor thyrotoxische PP. Episodische zwakte waargenomen bij thyrotoxische PP is vergelijkbaar met die waargenomen bij HypoPP en Andersen-Tawil syndroom. Deze aandoening komt het meest voor bij Aziaten en Latijns-Amerikaanse mannen. Thyrotoxic PP is een genetische aandoening ontmaskerd door thyrotoxicose. Kir2. 6 wordt voornamelijk uitgedrukt in skeletspieren. Triiodothyronine verbetert de transcriptie KCNJ18, die verbeterde uitdrukking van Kir2. 6 kan drijven. PKC wordt geactiveerd tijdens thyrotoxicose wegens verhoogde pip2 omzet en Kir kanalen direct interactie met PIP2 tijdens normale gating. Bij het syndroom van Andersen-Tawil is er een verminderde PIP2-affiniteit. In thyrotoxische PP verandert geen van de mutaties Kir2. 6 rectificatie.