Syndroom van Aicardi-Goutières: fenotypisch en genetisch spectrum in een reeks van drie gevallen | Anales de Pediatría
het syndroom van Aicardi-Goutières (AGS) is een zeldzame erfelijke ziekte waarvan de exacte prevalentie onbekend is. Het werd voor het eerst beschreven in 1984 door Jean Aicardi en Francoise Goutières als een progressieve encefalopathie met aanvang in de eerste levensmaanden gekarakteriseerd door cerebrospinale vloeistof (CSF) lymfocytose en calcificaties in de basale ganglia.1 Het manifesteert zich met prikkelbaarheid, psychomotorische retardatie, spasticiteit, dystonie, epileptische aanvallen, terugkerende episodes van aseptische koorts en microcefalie. De sterfte is hoger tijdens de encefalopathische fase, en hoewel de ziekte meestal daarna stabiliseert, veroorzaakt het ernstige neurologische gevolgen. Andere karakteristieke kenmerken die tijdens het verloop kunnen optreden zijn chilblains, oculaire symptomen (voornamelijk glaucoom), cardiale betrokkenheid of auto-immuunziekten.2 type I interferonen spelen een cruciale rol bij de pathogenese van AGS, waarbij hun expressie wordt gereguleerd, wat leidt tot een verhoogde productie.Daarom is een van de klassieke laboratoriumbevindingen bij deze patiënten een verhoogd interferon alfa-gehalte in de liquor cerebrospinalis, samen met pleiocytose en even hoge neopterine-en biopterinespiegels. Het potentiële nut van het beoordelen van de mate van expressie van interferon-gestimuleerde genen door interferon in perifeer bloed als marker wordt momenteel onderzocht, aangezien er aanwijzingen zijn dat deze niveaus hoog blijven na de encefalopathische fase (“interferon signature”).3-5 een ander belangrijk kenmerk is de detectie van neuroimaging afwijkingen met inbegrip van calcificaties in de basale ganglia en veranderingen in de witte stof (Fig. 1). Tot op heden kennen we 7 genen waarvan de mutaties kunnen leiden tot upregulatie van de interferonroute: ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1 en IFIH1. Heterozygote mutaties zijn beschreven voor de genen TREX1, ADAR en IFIH1, terwijl de mutaties in alle andere genen homozygote mutaties zijn.2 mutaties in het ifih1 gen werden het laatst gedetecteerd (2014)4 en zijn daarom de minst frequente pathogene varianten, terwijl mutaties in de genen RNASEH2B en TREX1 verantwoordelijk zijn voor het hoogste percentage gediagnosticeerde gevallen van AGS.
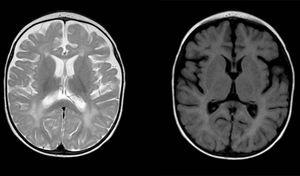
in de afgelopen decennia, dankzij de vooruitgang in de genetica die de detectie van deze specifieke mutaties mogelijk maakt, is er bewijs naar voren gekomen van een breed fenotypisch spectrum buiten de klassieke presentatie op basis van het causatieve gen. We presenteren de gevallen van 3 patiënten die in de afgelopen 8 jaar een diagnose van AGS hebben gekregen met als doel hun klinische kenmerken te analyseren in relatie tot het onderliggende genetische defect (Tabel 1). Over het algemeen waren de presenterende kenmerken van AGS consistent met die beschreven in de meest recente casusreeks in de literatuur: neonatale presentatie (33%), microcefalie (66%), psychomotorische retardatie (100%), spasticiteit (100%), ernstige verstandelijke beperking (66%) en calcificaties op craniale CT (66%), hoewel slechts één patiënt epileptische aanvallen had.
kenmerken van patiënten met het syndroom van Aicardi-Goutières.
| Geval 1 | Case 2 | Zaak 3 | |
|---|---|---|---|
| Genetica | Homozygote mutatie (blz.Ala177Thr) in RNASEH2B gen | Homozygote mutatie (341G>A) in TREX1 gen | Heterozygote mutatie (c.992C>G en p.Thr331Arg) in IFIH1 gen |
| Huidige leeftijd | 3 jaar | 7 jaar en 4 maanden | 12 jaar en 11 maanden |
| Seks | Man | Vrouw | Man |
| Herkomst | Roemenië | Spanje | Italië |
| AP | – | Week 36: intra-uteriene groei restrictie Week 37: microcefalie, verkalking van de placenta |
Gespleten gehemelte |
| Klinische verschijnselen | |||
| Leeftijd bij begin | 10 maanden | Geboorte – | 2 jaar |
| Eerste presentatie | Prikkelbaarheid Psychomotorische regressie |
Tremoren, hypotonie, zwak huilen, groeistoornis | Motorische vaardigheid vertraging |
| Psychomotorische retardatie | Ja | Ja | Ja |
| Taal | 2-Lettergreep words | No | Yes |
| Microcephaly | Yes | Yes | No |
| Chilblains | No | Yes | Yes |
| Epilepsy | No | Yes | No |
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFCS IV |
| bewegingsstoornissen | Geen | Ja | Geen |
| Abnormale oogbewegingen | Geen | Geen | Geen |
| Visual impairment | Geen | – | Bijziendheid |
| Glaucoom | Geen | Geen | Geen |
| gehoorverlies | – | – | Geen |
| Cardiale betrokkenheid | Geen | Milde tricuspidalisklep en de mitralisklep insufficiëntie | Geen |
| Terugkerende koorts | Geen | Geen | Geen |
| een verstandelijke handicap | Ja | Ja, graf | Ja, mild |
| Andere | – | – | Singleton-Merten syndroom: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
| CSF | |||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – |
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – |
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – |
| INF alfa (IU/mL) | 25 ( | 75 ( | – |
| Blood | |||
| Interferon signature | – | – | Gene overexpression |
| Neuroimaging | |||
| Calcifications on CT | No | Yes. In basale en periventriculaire ganglia | Ja. Symmetrische calcificaties in diepe WM van frontale regio en lentiform nucleus |
| hoofd MRI | Diffuse en fragmentarische veranderingen in WM intensiteit van beide cerebrale hemisferen, hyperintense op T2. Betrokkenheid van de subcorticale WM (het sparen van de U-vezels) en periventricular WM | Gegeneraliseerde WM betrokkenheid bij de verdwijning van de lobaire WM met inbegrip van de subcorticale U de vezels van de frontale, temporale en occipitale kwabben, bilateraal en symmetrisch, zonder corticale betrokkenheid | – |
GSK, cerebrospinale vloeistof; CT, computertomografie; GMFCS, Gross Motor Function Classification System; INF, interferon; IUGR, intra-uteriene groei restrictie; de MRI, magnetic resonance imaging; PNP, polyneuropathie; WM, wit mater.
zoals eerder opgemerkt, zijn de homozygote veranderingen in het gen RNASEH2B de frequentste varianten die AGS veroorzaken en hun fenotypic uitdrukking gewoonlijk het meest aan de klassieke presentatie in overeenstemming is.4 Dit was het geval voor de patiënt in onze studie die een dergelijke mutatie droeg, die begon op de leeftijd van 10 maanden met prikkelbaarheid en psychomotorische retardatie en met karakteristieke neuroimaging en CSF bevindingen.
twintig procent van de gevallen van AGS kan een neonatale presentatie hebben, waarbij de ziekte in utero begint.5 mutaties in een van de 7 bovengenoemde genen kunnen tot dit fenotype leiden, maar deze vroege presentatie wordt het vaakst geassocieerd met het Trex-gen.4,5 de initiële presentatie van deze vorm is vergelijkbaar met die van een FAKKELINFECTIE, met hepatosplenomegalie, hypertransaminasemie, trombocytopenie en neurologische manifestaties waaronder extreme prikkelbaarheid, bewegingsstoornissen en epileptische aanvallen.Deze patiënten hebben een ernstiger ziekteverloop en lopen een hoger risico op overlijden. De patiënt in ons monster dat gepresenteerd met een dergelijke variant had een neonatale presentatie en heeft momenteel de meest ernstige vorm van ziekte van de 3.
mutaties in het adar1-gen en in het bijzonder het ifih1-gen worden geassocieerd met een late aanvang van de symptomen, na 1 jaar leven met een normale psychomotorische ontwikkeling.5 in sommige van deze gevallen heeft het syndroom een goedaardig verloop met relatief behoud van taal en motorische vaardigheden. Onze patiënt met een mutatie in het ifih1 gen was een enkelvoudig geval omdat hij ook Singleton-Merten syndroom had, een zeldzame ziekte die ook veroorzaakt werd door een mutatie in het ifih1 gen en gekenmerkt werd door tanddysplasie, aorta calcificaties en osteoporose.6
ons doel is om de significante fenotypische variabiliteit van AGS en de associatie ervan met specifieke mutaties te onderstrepen met het oog op zowel het stimuleren van de overweging van deze diagnose in gevallen met presentaties die afwijken van de klassieke vorm van de ziekte als het leveren van aanvullende informatie over het verloop van de ziekte en de resultaten bij deze patiënten.