periodiske lammelser
en klinisk nyttig klassificering af primære periodiske lammelser, vist i tabel 1, inkluderer hypokalemiske, hyperkalemiske og paramyotoniske former.
tabel 1. Primær periodisk lammelse (modificeret fra Jurkat-Rott og Lehmann-Horn) (åben tabel i et nyt vindue)
|
sygdom |
Gene |
Protein |
arv |
Mutation |
|
HyperPP |
SCN4A |
Nav1.4 |
dominerende |
fint |
|
NormoPP |
fint (pore) |
|||
|
Paramyotoniacongenita |
fint |
|||
|
HypoPP type II |
fint (pore) |
|||
|
HypoPP for at |
CACNA1S |
Cav1.1 |
dominerende |
Gain (Kurt-pore) |
|
Thyrotoksicpp |
KCNJ18 |
Kir2.18 |
dominerende |
tab |
|
Andersens syndrom |
KCNJ2 |
Kir2.1 |
dominerende |
tab |
det fysiologiske grundlag for slap svaghed er uigennemtrængelighed af muskelmembranen (dvs.sarcolemma). Ændring af serumkaliumniveau er ikke den vigtigste defekt i primær PP; den ændrede kaliummetabolisme er et resultat af PP. I primær og thyrotoksisk PP forekommer slap lammelse med relativt små ændringer i serumkaliumniveauet, mens serumkaliumniveauer i sekundær PP er markant unormale.
ingen enkelt mekanisme er ansvarlig for denne gruppe af lidelser. Således er de heterogene, men deler nogle fælles træk. Svagheden er normalt generaliseret, men kan være lokaliseret. Kraniale muskler og åndedrætsmuskler spares normalt. Strækreflekser er enten fraværende eller formindsket under angrebene. Muskelfibrene er elektrisk uigennemtrængelige under angrebene. Muskelstyrke er normal mellem angreb, men efter et par år udvikles en vis grad af fast svaghed i visse typer PP (især primær PP). Alle former for primær PP (undtagen Becker myotonia congenita ) er enten autosomal dominerende arvet eller sporadisk (sandsynligvis som følge af punktmutationer).
spændingsfølsomme ionkanaler regulerer nøje generering af handlingspotentialer (korte og reversible ændringer af spændingen i cellulære membraner). Disse er selektivt og variabelt permeable ionkanaler. Energiafhængige iontransportører opretholder koncentrationsgradienter. Under genereringen af handlingspotentialer bevæger natriumioner sig over membranen gennem spændingsstyrede ionkanaler. Den hvilende muskelfibermembran polariseres primært ved bevægelse af chlorid gennem kloridkanaler og repolariseres ved bevægelse af kalium. Natrium -, chlorid-og calciumkanalopatier, som en gruppe, er forbundet med myotoni og PP. De funktionelle underenheder af natrium -, calcium-og kaliumkanaler er homologe. Natriumkanalopatier forstås bedre end calcium-eller chloridkanalopatier. Alle former for familiær PP viser den endelige mekanistiske vej, der involverer afvigende depolarisering, inaktivering af natriumkanaler og muskelfibers uigennemtrængelighed.
diskussion i denne artikel omhandler primært natrium -, calcium-og kaliumkanalopatier såvel som sekundære former for PP. Chloridkanalopatier er ikke forbundet med episodisk svaghed og diskuteres mere detaljeret i artiklerne om myotoniske lidelser.
resume af kanaldysfunktion i forskellige typer PP
med HyperPP hurtig kanalinaktivering er mutationer normalt placeret i de indre dele af transmembransegmenter eller i de intracellulære sløjfer, der påvirker dockingsstederne for den hurtige inaktiverende partikel, hvilket forringer hurtig kanalinaktivering, der fører til vedvarende Na+ strøm.
med HypoPP-hyperpolariseringsaktiveret kationlækage, der modvirker K+ -korrigeringsstrøm, forårsager mutationer yderste arginin-eller lysinsubstitution.
med NormoPP depolariseringsaktiveret kationlækage er mutationer i dybere placeringer af spændingsføler af domæne II ved codon R675.
ionkanaldysfunktion kompenseres normalt godt med normal ophidselse, og yderligere udløsere er ofte nødvendige for at producere muskeluønskelighed på grund af vedvarende membran depolarisering.
glukose-og kaliumindtag har de modsatte virkninger ved disse lidelser. I HyperPP udløser kaliumindtag angrebet, mens glukose forbedrer det. I modsætning hertil provokerer glukose hypokalemiske angreb, og kalium er behandlingen for angrebet.
Bemærk billedet nedenfor.
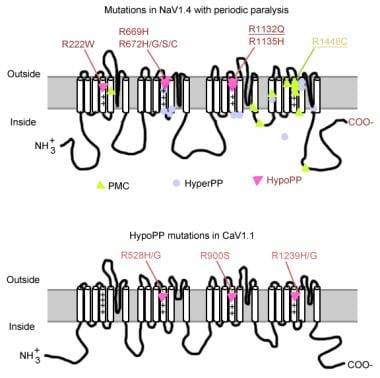 mutationer i periodisk lammelse.
mutationer i periodisk lammelse. muskelnatriumkanalgen
natriumkanalen har en alfa-underenhed og en beta-underenhed. Alfa-underenheden af natriumkanalen er et 260-kd glycoprotein omfattende omkring 1800-2000 aminosyrer. Denne kanal er stærkt bevaret evolutionært fra Drosophila til menneske. Det har 4 homologe Domæner (I-IV), der foldes for at danne en central pore, hver med 225-325 aminosyrer. Hvert domæne består af 6 hydrofobe segmenter (S1-S6), der krydser cellemembranen. Kanalens hovedfunktioner inkluderer spændingsfølsom gating, inaktivering og ionselektivitet. Den ekstracellulære sløjfe mellem S5 og S6 dypper ned i plasmamembranen og deltager i dannelsen af porerne. S4-segmentet indeholder positivt ladede aminosyrer i hver tredje position og fungerer som en spændingssensor. Konformationsændringer kan forekomme under depolarisering, hvilket resulterer i aktivering og inaktivering af kanalen. Den cellulære sløjfe mellem domæne III-S6 og domæne IV-S1 fungerer som en inaktiverende Port.
natriumkanalen har 2 porte (aktivering og inaktivering) og kan eksistere i 3 tilstande. I hvile med membranen polariseret lukkes aktiveringsporten, og inaktiveringsporten åbnes. Med depolarisering åbnes aktiveringsporten, så natriumioner kan passere gennem ionkanalen og også udsætte et dockingssted for inaktiveringsporten. Ved fortsat depolarisering lukker inaktiveringsporten, blokerer indgangen af natrium i cellen og får kanalen til at komme ind i hurtig inaktiveringstilstanden. Denne inaktivering af kanalen gør det muligt for membranen at blive repolariseret, hvilket resulterer i en tilbagevenden til hviletilstanden med aktiveringsporten lukket og inaktiveringsporten åbnet. To inaktiveringsprocesser forekommer i pattedyrs skeletmuskulatur: hurtig inaktivering involverer afslutning af handlingspotentialet og virker på en millisekund tidsskala. Langsom inaktivering tager sekunder til minutter og kan regulere populationen af spændende natriumkanaler.
Natriumkanalmutationer, der forstyrrer hurtig og langsom inaktivering, er normalt forbundet med en fænotype af HyperPP og myotoni, hvor som mutationer, der forbedrer langsom eller hurtig inaktivering, der producerer tab af natriumkanalfunktion, forårsager HypoPP.
mutationer af natriumkanalgenet (SCN4A) har flere generelle træk. De fleste af mutationerne er i den “inaktiverende” linker mellem gentagelser III og IV, i “spændingsfølende” segment S4 af gentagelse IV eller ved den indre membran, hvor de kan forringe dockingsstedet for inaktiveringsporten. Den kliniske fænotype adskiller sig ved specifik aminosyresubstitution, og mens der kan forekomme en vis overlapning mellem hyperkalæmisk PP, paramyotonia congenita (PC) og kaliumforværrede myotonier (PAM), er de 3 fænotyper generelt forskellige (som beskrevet nedenfor). Næsten alle mutante kanaler har nedsat hurtig inaktivering af natriumstrøm. De fleste patienter er følsomme over for systemisk kalium eller kold temperatur.
der findes to populationer af kanaler, mutant og vildtype; den nedsatte hurtiginaktivering resulterer i langvarig depolarisering af de mutante muskelfibermembraner og kan forklare de 2 kardinale symptomer på disse lidelser, myotoni og svaghed. I hyperkalæmisk PP forekommer en forstærkning af funktion i mutantkanalgating, hvilket resulterer i en øget natriumstrøm, der overdrevent depolariserer den berørte muskel. Mild depolarisering (5-10 mV) af myofibermembranen, som kan være forårsaget af øgede ekstracellulære kaliumkoncentrationer, resulterer i, at de mutante kanaler opretholdes i den ikke-aktiverede tilstand. Den vedvarende indadgående natriumstrøm forårsager gentagen affyring af vildtypens natriumkanaler, som opfattes som stivhed (dvs.myotoni).
hvis der er en mere alvorlig depolarisering (20-30 mV), fastgøres både normale og unormale kanaler i en tilstand af inaktivering, hvilket forårsager svaghed eller lammelse. Således kan subtile forskelle i sværhedsgraden af membran depolarisering gøre forskellen mellem myotoni og lammelse. Temperaturfølsomhed er et kendetegn for PC. Kold forværrer myotoni og inducerer svaghed. En række mutationer er forbundet med denne tilstand, 3 af dem på samme sted (1448) i S4-segmentet. Disse mutationer erstatter arginin med andre aminosyrer og neutraliserer denne stærkt konserverede S4-positive ladning. Mutationer af disse rester er den mest almindelige årsag til PC. Nogle af de mulige mekanismer, der er ansvarlige for temperaturfølsomhed, omfatter følgende:
-
temperatur kan differentielt påvirke konformationsændringen i mutantkanalen.
-
lavere temperaturer kan stabilisere mutantkanalerne i en unormal tilstand.
-
mutationer kan ændre kanalens følsomhed over for andre cellulære processer, såsom phosphorylering eller anden budbringere.
de fleste tilfælde af hyperkalæmisk PP skyldes 2 mutationer i SCN4A, T704M og M1592V. Mutationer i natriumkanalen, især ved rester 1448 og 1313, er ansvarlige for paramyotonia congenita. En lille del af hypokalemiske periodiske lammelsestilfælde er forbundet med mutationer ved kodoner 669 og 672 (HypoPP2). I HypoPP2 forbedrer natriumkanalmutationer inaktivering for at producere et nettotab af funktionsdefekt.
Normokalemisk PP ligner både HyperPP (kaliumfølsomhed) og HypoPP (varighed af angreb) og er forårsaget af SCN4A-mutationer på en dybere placering af spændingsføler DII ved codon 675. R675-mutationer adskiller sig fra HypoPP ved, at disse mutationer resulterer i depolariseringsaktiveret gating pore, der genererer kursstrøm med omvendt spændingsafhængighed, da dette sted udsættes for ekstracellulære steder ved stærkere depolarisering.
Calciumkanalgen
calciumkanalgenet (CACNL1A3) er et kompleks af 5 underenheder (alfa-1, alfa-2, beta, gamma og delta). Skeletmuskulaturdihydropyridin (DHP) – receptoren er primært placeret i den tværgående rørformede membran. Alpha – 1-underenheden har bindingssteder for DHP-lægemidler og udfører den langsomme L-type calciumstrøm. Det deltager også i eksitation-sammentrækning (EC) kobling og fungerer som en spændingssensor gennem dens kobling med ryanodinreceptoren af sarkoplasmatisk retikulum (dvs.calciumfrigivelseskanal). Eventuelle ændringer i membranpotentialet er forbundet med intracellulær calciumfrigivelse, hvilket muliggør EC-kobling. Punktmutationer i DHP-receptor / calciumkanal alfa-1-underenhed forårsager hypokalæmisk PP (HypoPP1). To mutationer af cacna1s gen, R528H og R1239H, er ansvarlige for de fleste tilfælde af hypokalæmisk PP.
det fysiologiske grundlag for sygdom forstås stadig ikke, men skyldes mere sandsynligt en svigt i ophidselse snarere end en svigt i EC-kobling. Imidlertid kan hypokalæmi-induceret depolarisering reducere calciumfrigivelse, hvilket påvirker spændingskontrollen af kanalen direkte eller indirekte gennem inaktivering af natriumkanalen. Insulin og adrenalin kan virke på en lignende måde. Mutationer af calciumkanalgenet har nogle ligheder med SCN4A-mutationer. Mutationer ændrer kanalinaktivering, men ikke spændingsafhængig aktivering. Optagelser fra myotube-kulturer fra berørte patienter afslørede en 30% reduktion i den DHP-følsomme L-type calciumstrøm. Kanaler inaktiveres ved lave membranpotentialer.
Calciumkanalmutationer forårsager et tab af funktion manifesteret som en reduceret strømtæthed og langsommere inaktivering. Hvordan denne inaktivering er relateret til hypokalæmi-inducerede angreb forstås ikke. I det mindste i R528H-mutation forekommer en mulig sekundær kanalopati, bundet til en reduktion i den ATP-følsomme kaliumstrøm fra ændret calciumhomeostase. De lavere strømme forbundet med cacnl1a3-mutationer kunne let ændre intracellulær calciumhomeostase, hvilket kunne påvirke egenskaberne og ekspressionen af K+ – kanaler, især KATP (ATP-følsom kaliumkanal), der tilhører indadgående ensretterklasse af kanaler. Insulin virker også i HypoPP ved at reducere denne indadgående ensretter K+ strøm.
Spændingssensorladningstab tegner sig for de fleste tilfælde af HypoPP. Natrium – og calciumkanaler har homologe poredannende alfa-underenheder. Punktmutationer i CACNL1A3 og SCN4A påvirker argentinske rester i S4-spændingssensorerne på disse kanaler. Argininmutationer i S4-segmenter er ansvarlige for 90% af HypoPP-tilfælde.
Spændingssensorladningstab tegner sig for de fleste tilfælde af HypoPP. Natrium – og kalciumkanaler har homologe poredannende underenheder. Næsten alle mutationerne i Cav1.1 (HypoPP-1) og Nav1.4 (HypoPP-2) neutraliserer en positivt ladet aminosyre i en af de yderste argininer eller lysiner af spændingssensorer. Nav1.4 mutationer er oftest placeret i spændingssensorerne i I, II og III gentagelser, hvilket forårsager en kationlækage.
Substitution af yderste arginin med en mindre aminosyre, såsom glycin, åbner en ledende vej ved hyperpolariseret potentiale, hvilket resulterer i en indadvendt kationstrøm (kationlækage eller kursstrøm at skelne fra (kur-) gennem ionledende pore, er en hyperpolariseringsaktiveret strøm af monovalente kationer gennem S4 gating pore modvirker korrigering af K+ strømme) depolariserende eller destabiliserende hvilepotentialet.
S4-segmentet bevæger sig udad under depolarisering, der lukker den ledende vej. Muskelfibre med svære spændingsfølermutationer depolariseres ikke kun under hypokalæmi, men også ved kaliumniveauer i det normale interval, hvilket forklarer interictal og permanent svaghed. Alvorlig myopati med fedtudskiftning af muskelvæv findes almindeligvis hos patienter med Cav1.1 R1239H (DIV-mutationer).
glukokortikosteroider forårsager HypoPP ved at stimulere Na+ K+ ATPase medieret af insulin og amylin.
Kaliumkanalgen
indadgående rettelse er en vigtig egenskab ved Kir-kanaler. Berigtigelse involverer spændingsafhængig ledningsafhængig poreblokering af pore med polyaminer og Mg++ under depolarisering, og denne blokering fjernes under potentiel gradient under hyperpolarisering. Kaliumkanalmutationer ses ved Andersen-Tavils syndrom og thyrotoksisk PP.
triaden af dysmorfe træk, periodisk lammelse og hjertearytmi karakteriserer Andersen-Tavils syndrom. Dette syndrom er forbundet med mutationer i KCNJ2-genet. KCNJ2-genet koder for den indadrettede kaliumkanal Kir2.1. Kaliumkanalmutationer i KCNE3 er rapporteret at forårsage hypokalæmisk PP, men dette er ikke dokumenteret.
mutationer i Kir2.6 forårsager modtagelighed for thyrotoksisk PP. Episodisk svaghed set i thyrotoksisk PP svarer til den, der ses i HypoPP og Andersen-Tavil syndrom. Denne lidelse er mest udbredt hos asiater og latinamerikanske mænd. Thyrotoksisk PP er en genetisk lidelse, der ikke er maskeret af thyrotoksikose. Kir2. 6 er primært udtrykt i skeletmuskulatur. Triiodothyronin forbedrer kcnj18 transkription, som kan drive forbedret ekspression af Kir2.6. PKC aktiveres under thyrotoksikose på grund af øget PIP2-omsætning, og Kir-kanaler interagerer direkte med PIP2 under normal gating. Ved Andersen-Tavils syndrom er der nedsat PIP2-affinitet. I thyrotoksisk PP ændrer ingen af mutationerne Kir2.6-rettelse.