Edukacja mózgowa
chociaż każdego dnia dowiadujemy się więcej o patofizjologii choroby Parkinsona, nadal jest ona uważana za w dużej mierze idiopatyczną (o nieznanej przyczynie). Prawdopodobnie wiąże się to z interakcją podatności żywiciela i czynników środowiskowych. Niewielki odsetek przypadków jest uwarunkowany genetycznie, a czynniki genetyczne są intensywnie badane.
fizjologicznie objawy związane z chorobą Parkinsona są wynikiem utraty wielu neuroprzekaźników, przede wszystkim dopaminy. Objawy nasilają się z czasem, ponieważ coraz więcej komórek dotkniętych chorobą jest traconych. Przebieg choroby jest bardzo zmienny, niektórzy pacjenci wykazują bardzo niewiele objawów w miarę starzenia się, a inni, których objawy postępują szybko.
choroba Parkinsona jest coraz częściej postrzegana jako złożona choroba neurodegeneracyjna z sekwencją progresji. Istnieją silne dowody na to, że najpierw wpływa na grzbietowe jądro motoryczne nerwu błędnego i cebulki węchowe i jądro, następnie locus coeruleus, a ostatecznie substantia nigra. Korowe obszary mózgu są dotknięte w późniejszym etapie. Uszkodzenia tych różnych systemów neuronalnych odpowiadają za wieloaspektowe zmiany patofizjologiczne, które powodują upośledzenie nie tylko układu ruchowego, ale także systemu poznawczego i neuropsychologicznego (Kwan & Whitehill, 2011).
rola dopaminy
dopamina, podobnie jak inne neuroprzekaźniki, przekazuje wiadomości chemiczne z jednej komórki nerwowej do drugiej przez synapsę, przestrzeń między komórką presynaptyczną a receptorem postsynaptycznym. Dopamina jest wydzielana do synapsy z pęcherzyków magazynujących błonę w błonie presynaptycznej. Przenika przez synapsę i wiąże się z błoną postsynaptyczną, gdzie aktywuje receptory dopaminy. Niewykorzystana dopamina pozostała w synapsie jest wchłaniana z powrotem do komórki presynaptycznej; po powrocie do komórki presynaptycznej nadmiar dopaminy jest pakowany do pęcherzyków magazynujących i ponownie uwalniany do synapsy.
w synapsie, gdy dopamina przemieszcza się z jednej komórki do drugiej, może zostać rozbita i unieczynniona przez dwa enzymy, MAO (oksydaza monoaminowa) i COMT (katechol-o-metylotransferaza). Jedna ze strategii terapeutycznych wprowadza do synapsy inhibitor MAO, który przerywa działanie enzymu MAO i zapobiega rozpadowi dopaminy. To pozwala więcej dopaminy pozostać w synapsie i zwiększa prawdopodobieństwo, że będzie wiązać się z błoną postsynaptyczną.
chemiczna Transmisja synaptyczna
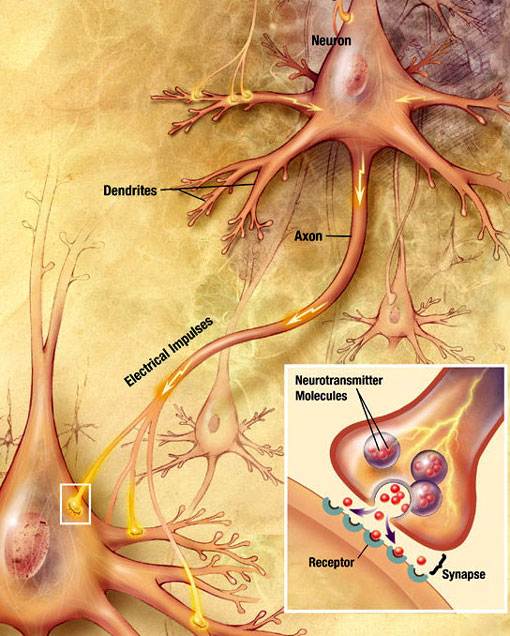
fala elektrochemiczna zwana potencjałem czynnościowym przemieszcza się wzdłuż aksonu neuronu. Gdy potencjał czynnościowy dociera do terminala presynaptycznego, wywołuje uwalnianie niewielkiej ilości cząsteczek neuroprzekaźników, które wiążą się z cząsteczkami receptora chemicznego zlokalizowanymi w błonie neuronu postsynaptycznego, po przeciwnej stronie rozszczepu synaptycznego. Źródło: Wikimedia Commons.
postępująca utrata dopaminy
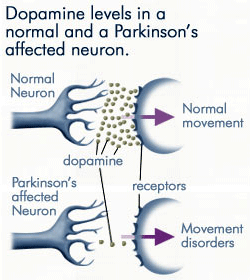
ponieważ neurony dotknięte chorobą Parkinsona wytwarzają coraz mniej dopaminy, znacznie mniej dopaminy jest dostępnych do wiązania się z receptorami dopaminy na błonie postsynaptycznej. Źródło: anti-agingfirewalls.com.
chociaż utrata komórek dopaminy nie może być mierzona bezpośrednio, pomiary u zdrowych neurologicznie ludzi i naczelnych nieludzkich ujawniają powolną postępującą utratę dopaminy wraz z wiekiem. W chorobie Parkinsona utrata występuje w znacznie większym tempie, a zarówno środki biochemiczne, jak i badania obrazowe sugerują, że istnieje znaczny spadek dopaminy w czasie pojawienia się objawów motorycznych. W tym ujęciu choroba Parkinsona jest przyspieszoną wersją śmierci komórki obserwowanej przy normalnym starzeniu się (Cookson, 2009). Jest to zilustrowane na poniższym wykresie, który pokazuje spadek neuronów dopaminergicznych podczas normalnego starzenia się, w idiopatycznej PD, w PD spowodowanej czynnikami środowiskowymi lub genetycznymi oraz we wczesnym początku PD.
Ewolucja zubożenia dopaminy w chorobie Parkinsona
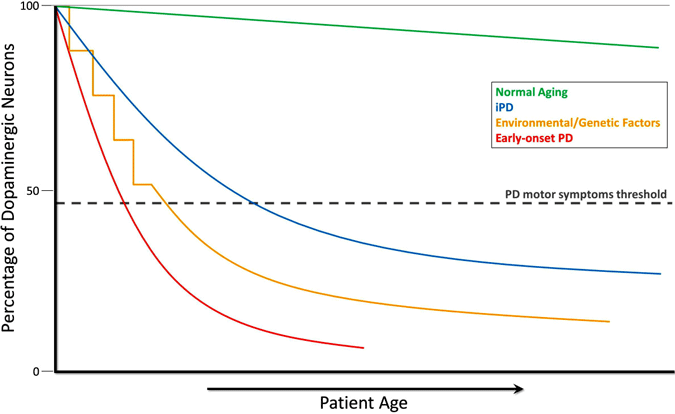
podczas normalnego starzenia (zielona linia), małe, ale powolne zwyrodnienie dopaminergiczne występuje bez żadnych objawów motorycznych. Idiopatyczna PD (IPD, Niebieska linia) jest nieznanego pochodzenia, ale uważa się, że rozwija się stopniowo, z powolną degeneracją neuronów dopaminergicznych prowadzącą do klasycznych objawów ruchowych PD w późniejszym życiu. Inny model neurodegeneracji dopaminy prowadzący do objawów ruchowych PD obejmuje powtarzające się narażenie na toksyczne czynniki środowiskowe w czasie w połączeniu z genetyczną predyspozycją do utraty neuronów dopaminergicznych (żółta linia). Wczesny początek PD (czerwona linia), spowodowany mutacjami w genie Parkina, wiąże się z gwałtownym spadkiem neuronów dopaminergicznych, a objawy ruchowe PD mogą występować dziesiątki lat wcześniej niż te w idiopatycznej PD. Jeszcze jeden scenariusz (nie pokazany)rozwoju objawów ruchowych PD obejmuje możliwe toksyczne czynniki środowiskowe w macicy lub czynniki genetyczne prowadzące do Nietypowo niskiej liczby neuronów dopaminergicznych przy urodzeniu i zwiększonej podatności na rozwój PD (Haas i wsp ., 2012).
degeneracja neuronów dopaminy jest szczególnie widoczna w części substancji czarnej zwanej Pars compacta. Co istotne, utrata dopaminy w Pars compacta zwiększa ogólny napęd pobudzający w zwojach podstawnych, * zakłócając dobrowolną kontrolę silnika i powodując charakterystyczne objawy PD. Normalizację funkcji motorycznych obserwuje się początkowo po leczeniu lewodopą (Gasparini i wsp ., 2013).
*głównymi składnikami zwojów podstawy są prążkowie (jądro ogoniaste i skorupa), globus pallidus, substantia nigra, jądro półleżące i jądro subtalamiczne.
wraz ze wzrostem nasilenia PD, wyczerpanie dopaminy prowadzi do dalszych zmian w ścieżkach zwojów podstawnych, w tym zmiany funkcji innych neuroprzekaźników zwojów podstawnych, takich jak glutaminian, GABA i serotonina (Gasparini i in., 2013). Chociaż istnieje względna podatność neuronów produkujących dopaminę w istocie czarnej, nie wszystkie komórki dopaminy są dotknięte chorobą Parkinsona; w niektórych częściach mózgu neurony produkujące dopaminę są stosunkowo oszczędzone (Cookson, 2009).
szlak Nigrostriatalny
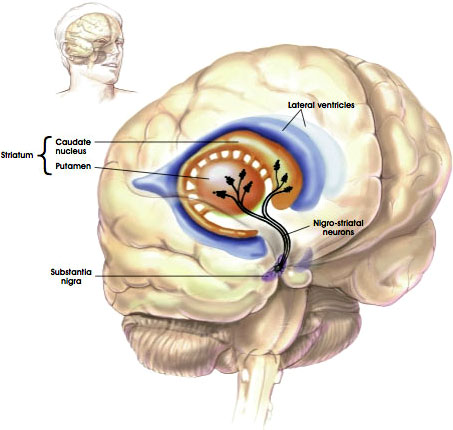
.
ciała Lewy 'ego i alfa-synukleina
ciała Lewy’ ego są nieprawidłowymi agregatami i inkluzjami białka, które rozwijają się wewnątrz komórek nerwowych u osób z chorobą Parkinsona. Agregacje zwykle składają się z nierozpuszczalnych agregatów włóknistych zawierających źle złożone białka. Duża liczba cząsteczek została zidentyfikowana w ciałach Lewy ’ ego, ale głównym składnikiem jest białko zwane Alfa-synukleiną.
ciała Lewy (inkluzje Alfa-Synukleinowe)
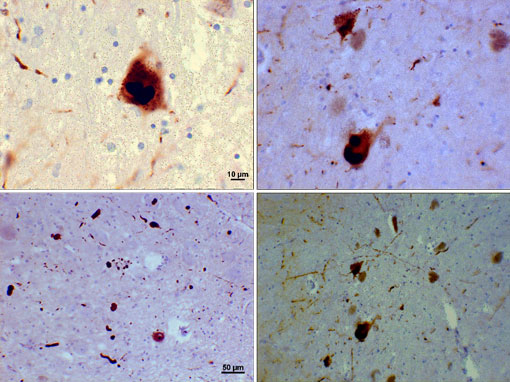
Fotomikrograf regionów substantia nigra u pacjenta z chorobą Parkinsona pokazujący ciała Lewy 'ego i nerwy Lewy’ ego w różnych powiększeniach. Górne panele pokazują 60-krotne powiększenie inkluzji wewnątrznaronowych Alfa-synuklein połączonych w ciała Lewy ’ ego. Dolne panele to obrazy o powiększeniu 20×, które pokazują neuryty lewe podobne do nici i zaokrąglone ciała lewe o różnych rozmiarach. Zdjęcia dzięki uprzejmości Suraj Rajan.
patologia Lewy 'ego obejmuje wiele regionów mózgu, a niektóre raporty sugerują, że substancja Czarna nie jest pierwszym miejscem, w którym tworzą się ciała Lewy’ ego w chorobie Parkinsona. Inkluzje i skupiska prawdopodobnie symbolizują końcową fazę kaskady skomplikowanych zdarzeń. Wcześniejszy etap może być bardziej bezpośrednio związany z patogenezą zaburzenia niż same inkluzje, które mogą lub nie mogą reprezentować cechy diagnostyczne.
ciała Lewy ’ ego są również widoczne w „otępieniu z ciałami Lewy 'ego”, co sugeruje, że Warunki te są ze sobą powiązane przez wspólną patologię i prawdopodobnie przez wspólną etiologię. Ani utrata komórek, ani tworzenie ciał Lewy ’ ego nie są absolutnie specyficzne dla PD, ale oba są wymagane do diagnozy PD zgodnie z aktualnymi definicjami (Cookson, 2009).
zaburzenia neurodegeneracyjne, takie jak choroba Alzheimera, degeneracja czołowo-skroniowa, choroba prionowa, pląsawica Huntingtona i choroby motoneuronowe coraz częściej zdają sobie sprawę, że mają wspólne mechanizmy komórkowe i molekularne, w tym agregację białek i tworzenie ciała inkluzji w niektórych obszarach układu nerwowego (Jellinger, 2011).
stan zapalny i odpowiedź immunologiczna
wydaje się, że przyczyną zwyrodnienia dopaminergicznego jest wieloczynnikowy wpływ zarówno elementów endogennych, jak i środowiskowych. Zapalenie i odpowiedź immunologiczna są coraz częściej uważane za ważne mediatory zwyrodnienia dopaminergicznego. Duże badania populacyjne sugerują, że osoby przyjmujące niesteroidowe leki przeciwzapalne (NLPZ) mają mniejsze ryzyko wystąpienia idiopatycznej PD, co sugeruje, że leki przeciwzapalne mogą być obiecującym leczeniem modyfikującym przebieg choroby u pacjentów z chorobą Parkinsona (Barcia, 2013).
nowe fazy badań obejmowały leczenie przeciwzapalne-w szczególności szukanie obiektywnego biomarkera w leczeniu mającym na celu zmniejszenie zmian zapalnych u pacjentów z PD. Naukowcy używają narzędzi neuroobrazowania do opracowania odpowiedniego biomarkera z zamiarem przetestowania go w dużych badaniach obrazowania klinicznego. Wyniki tych badań dostarczą danych do testowania i monitorowania postępu leczenia przeciwzapalnego PD i pomogą zidentyfikować terminowe okno terapeutyczne, aby zatrzymać lub przynajmniej spowolnić zapalne zwyrodnienie dopaminergiczne (Barcia, 2013).
parkinsonizm
parkinsonizm, znany również jako „atypowy Parkinson”, „wtórny Parkinson” lub „zespół Parkinsona” jest zespołem neurologicznym, w którym pacjent wykazuje niektóre objawy związane z chorobą Parkinsona—drżenie, sztywność, bradykineza i niestabilność postawy. Ale parkinsonizm nie jest chorobą Parkinsona. Uważa się, że parkinsonizm nie jest spowodowany chorobą Parkinsona, a pacjenci zazwyczaj słabo reagują na interwencję farmakologiczną. Parkinsonizm często ma identyfikowalną przyczynę, taką jak ekspozycja na toksyny, metamfetaminę, uraz, wielokrotne udary, inne zaburzenia układu nerwowego lub choroba. Ogólnie rzecz biorąc, ciała lewe nie są widoczne w parkinsonizmie.
termin parkinsonizm jest również związany z zaburzeniami, takimi jak postępujące porażenie nadjądrowe, atrofia wielu układów, otępienie ciała lewego, zwyrodnienie korowo-nosowe, parkinsonizm naczyniowy, parkinsonizm indukowany lekami i parkinsonizm wtórny do infekcji i innych przyczyn (Hohler et al., 2012). Postać odwracalnego parkinsonizmu może wystąpić podczas stosowania niektórych leków neuroleptycznych, w szczególności rezerpiny, leków przeciwpsychotycznych (haloperydolu) i metoklopramidu. Narażenie na niektóre toksyny, ciężkie zatrucie tlenkiem węgla i zatrucie rtęcią może również prowadzić do parkinsonizmu.
pojawienie się na początku lat 80. objawów parkinsonizmu u grupy narkomanów, którzy spożywali zanieczyszczoną partię syntetycznego opiatu, doprowadziło do odkrycia chemicznego MPTP jako czynnika, który powoduje zespół parkinsonizmu u naczelnych nieludzkich, jak również u ludzi. MPTP może być wytwarzany podczas wytwarzania postaci heroiny (MPTP jest przekształcany w neurotoksynę, która selektywnie niszczy komórki dopaminy w substantia nigra). Przypadki te są rzadkie i dotyczą głównie długotrwałych osób zażywających narkotyki.
nadużywanie metamfetaminy jest również związane z parkinsonizmem. U zwierząt doświadczalnych ekspozycja na metamfetaminę uszkadza włókna dopaminergiczne w prążkowiu*, a także ciała komórkowe w istocie czarnej, odzwierciedlając zwyrodnienie obserwowane u ludzi z PD. Selektywne uszkodzenie terminali dopaminergicznych w prążkowiu zaobserwowano również u ludzi zażywających metamfetaminę, chociaż do tej pory nie ma dowodów na to, że nadużywanie metamfetaminy uszkadza ciała komórek dopaminergicznych w substantia nigra (Granado et al., 2013).
* największe jądro zwojów podstawnych, prążkowie składa się z jądra ogoniastego i skorupy.
postawiono hipotezę, że stosowanie metamfetaminy może predysponować użytkowników do przyszłego rozwoju PD. Hipoteza ta została poparta przez ostatnie prace epidemiologiczne wskazujące, że użytkownicy metamfetaminy mają zwiększone ryzyko rozwoju PD. Jest to zgodne z utrzymującymi się neurotoksycznymi skutkami metamfetaminy u zwierząt doświadczalnych(Granado et al., 2013).
pacjenci z parkinsonizmem są często trudni do opanowania jako pacjenci ambulatoryjni. Złożoność ich objawów, dodatkowe deficyty poznawcze i autonomiczne, słaba reakcja na większość leków PD i stosunkowo szybki spadek stanu przyczyniają się do wyzwań w zarządzaniu tymi pacjentami, szczególnie w miarę postępu choroby (Hohler et al., 2012).