Ginekologia i położnictwo opis przypadku
słowa kluczowe
Tetra-amelia; malformacja; genotyp; fenotyp.
wprowadzenie
anomalie kończyn stanowią ważną grupę wrodzonych patologii charakteryzujących się hipoplazją lub całkowitą aplazją jednej lub więcej kości kończyny. Nieprawidłowości kończyn wszystkich typów występują u około 1 na 1300 do 2000 urodzeń. Te anomalie kończyn mogą być izolowane lub związane z innymi anomaliami . Zespół tetraamelii jest rzadki i pozostają szare obszary.
zgłaszamy dwa przypadki tetra-Amelii w położnictwie II stopnia w Dakarze (Senegal), które są podobne do tetraamelii-1 (chromosom 17q21), tetraamelii-2 (chromosom 8q23) i zespołu Roberta (chromosom 8p21). To ilustruje trudności w korelacji fenotypu i genów zaangażowanych.
opisy przypadków
Przypadek 1
Pani AD była 44-letnią matką skierowaną do naszego oddziału w 36 tygodniu ciąży z poważnymi anomaliami przedrzucawkowymi i płodowymi. Miała pięć lat bez żadnych anomalii płodowych. Teraz nie paliła i nigdy nie paliła ani nie piła alkoholu. Nie była narażona na bierne palenie. Była w związku małżeńskim trzeciego stopnia dla wszystkich swoich dzieci. Ms. AD miał negatywny wynik na wirusowe zapalenie wątroby typu B, HIV i kiłę. Była chroniona przed wirusem różyczki i nie miała wcześniejszej ekspozycji na Toxoplasma gondii. Badanie USG wykonywane pod koniec 33 tygodnia i 35 tygodnia ciąży wykazało oligoamniozę i wodogłowie oraz agenezę kończyn. Recepty w czasie ciąży obejmowały podawanie żelaza i kwasu foliowego, a także podawanie pirymetaminy sulfadoksyny. Ten ostatni przepisywano w 18. tygodniu, a następnie w 26. tygodniu w ramach profilaktyki przeciw malarii u kobiet w ciąży. Wysokość symphyseal-fundal mierzona 28 cm. Z powodu ciężkich cech stanu przedrzucawkowego; została natychmiast hospitalizowana i obserwowana w jednostce porodowej i porodowej. Następnie początkowo otrzymała IV siarczan magnezu, aby zapobiec rzucawce i leki przeciwnadciśnieniowe, aby utrzymać skurczowe ciśnienie krwi poniżej 160 mmHg i rozkurczowe ciśnienie krwi poniżej 105 mmHg.
zapadła decyzja o natychmiastowym cesarskim cięciu. Wyekstrahowano 2150 gramów żywych urodzonych, które następnie zmarły w ciągu 10 minut. Przed śmiercią ciało przedstawiało ruchy pełzające. Zidentyfikowano kilka anomalii zewnętrznych (ryc. 1), w tym całkowitą agenezę wszystkich czterech kończyn, wodogłowie o obwodzie głowy 39 cm. Na twarzy pojawił się hiperteloryzm z kolobomą powiek, łagodnym egzoftalmosem i aniridią. Usta były odwrócone V bez wyraźnego rozgraniczenia warg, a nos był szczątkowy. Noworodek był pozbawiony skóry (włosów i brwi). Uszy zostały zredukowane do szkiców, które wyglądały jak szczeliny. Szyja była krótka. Tułów zredukowano do 26-centymetrowej stożkowatej struktury z pępowiną na dolnym końcu. Klatka piersiowa była wąska. Tuż poniżej pępka tułów poszerzony do tyłu znajdował się na dnie słupa ogonowego, linii środkowej z recesjami i pączkami, które mogły odpowiadać fallusowi nieokreślonego rodzaju. Stwierdzono agenezę miednicy, narządów płciowych i imperforacji odbytu. Nie przeprowadzono patologii płodu. Jednak śmierć w ciągu 10 minut po porodzie i stożkowy wygląd klatki piersiowej może sugerować nieprawidłowości płuc.
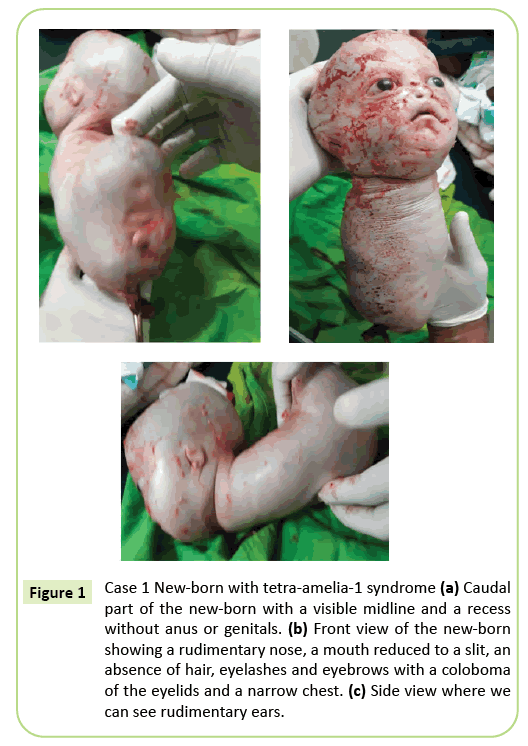
Rysunek 1: Przypadek 1 noworodek z zespołem tetra-amelia-1 (a) część ogonowa noworodka z widoczną linią środkową i wgłębieniem bez odbytu i narządów płciowych. b) Widok Z Przodu noworodka pokazujący prymitywny nos, usta zredukowane do szczeliny, brak włosów, rzęs i brwi z kolobomą powiek i wąską klatką piersiową. (c) widok z boku, gdzie widzimy prymitywne uszy.
Przypadek 2
drugim przypadkiem była 22-letnia primigravida skierowana do naszej placówki na badanie USG w 37 tygodniu ciąży. Nie była w związku małżeńskim. Miała negatywny wynik na wirusowe zapalenie wątroby typu B, HIV i kiłę. Nie była badana na toksoplazmozę i różyczkę. Podczas ciąży nie przeprowadzono badań USG. Badanie kliniczne wykazało opóźnienie wzrostu płodu (wysokość Fundal: 26 cm). Wyniki badań ultrasonograficznych wykazały, że kość ramienna jest zniekształcona, mierząc 23,9 mm, co odpowiada 17 tygodniom ciąży. Była ageneza kości udowej. Skrzydła biodrowe były widoczne na USG. Nie stwierdzono nieprawidłowości w płucach i sercu. Dostawa została rozpoczęta. Noworodek miał fenotyp żeński z wynikiem Apgar 9 W 5. minucie. Morfologia głowy i tułowia nie była szczególna. Kończyny górne zostały zredukowane do dwóch pni o długości 3 cm. Stwierdzono całkowitą agenezę 2 kończyn dolnych. Była to symetryczna anomalia (ryc. 2).
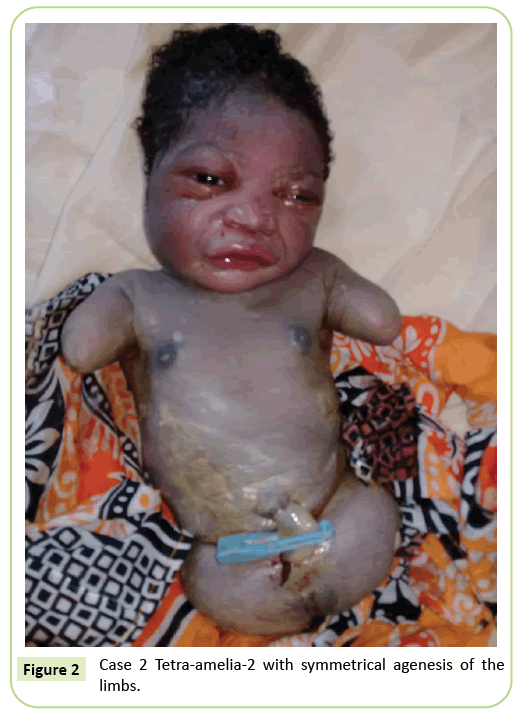
Rysunek 2: Przypadek 2 Tetra-amelia-2 z symetryczną agenezą kończyn.
dyskusja
opisał w 2011 epidemiologię wad wrodzonych na podstawie danych zebranych z 20 programów nadzoru wad wrodzonych ze wszystkich kontynentów z wyjątkiem Afryki w latach 1968-2006. W sumie zidentyfikowano 326 przypadków Amelii wśród 23 110 591 żywych urodzeń, martwych urodzeń i aborcji. Częstość występowania wynosiła 1,41/100 000 .
Tetra-amelia odnosi się do całkowitego braku kończyn i występuje rzadziej. Według naszej wiedzy tetra-amelia-1 jest opisana w 7 rodzinach. Wygląda na dziedziczenie autosomalne recesywne. We wszystkich rodzinach tetra-amelia-1 była związana z poważnymi wadami rozwojowymi innych części ciała, w tym twarzy i głowy, anomaliami układu nerwowego, szkieletu i narządów płciowych. Płuca były w wielu przypadkach słabo rozwinięte, co utrudnia lub uniemożliwia oddychanie . Zimmer i in. opisano w 1985 roku silnie wsobną rodzinę, w której 6 niemowląt miało tetra-1 i wodogłowie. Opisali u jednego z płodów całkowity brak kości miednicy, rozszczep wargi i podniebienia, arrhinię i aplazję uszu. Odnotowano również obustronne lewe płuco, uporczywy kanał tętniczy, imperforację odbytu. Badania płodu wyeliminowały diagnozę zespołu Roberta . Inne przypadki Znalezione w literaturze obejmują Kosaki et al., w 1996, z płodu kariotypu 46, XX z tetrafokomelią i ciężką hipoplazją płuc, oprócz anomalii twarzy i głowy . Rosenak i in. opisał przypadek tetra-Amelii z ciężką hipoplazją płuc u dwóch płodów pary nie spokrewnionej. Badania płodu wykluczyły diagnozę zespołu Roberta . Dwa dodatkowe przypadki zostały zgłoszone przez Złotogora i wsp. w 1993 roku. Obaj pacjenci zmarli wkrótce po urodzeniu, a autorzy sugerowali istnienie hipoplazji płuc. Niemann et al. zgłosiła pokrewieństwo Tureckiej rodziny, w której 4 z 8 braci cierpiało na tetra-Amelię. Oprócz braku 4 kończyn, badania płodowe 3 płodów ujawniły wiele anomalii: rozszczep warg i /lub podniebienia, laparoschisis, anomalie płucne, hipoplazja miednicy, atrezja choanas, pochwa i imperforacja odbytu . Wreszcie, w 2005, Krahn et al. opisano 2 braci urodzonych przez wsobnych rodziców cierpiących na tetraamelię i ciężką hipoplazję płuc. Obojczyki i łopatki były w normie u drugiego płodu. Kariotyp był normalny .
zespół Tetra-Amelii-1 lub TETAMS1 jest spowodowany homozygotyczną mutacją w genie WNT3 na chromosomie 17Q21 z dziedziczeniem autosomalnym recesywnym. Zespół tetraamelii – 2 (TETAMS2) charakteryzuje się szczątkowymi kończynami lub całkowitym brakiem kończyn, zazwyczaj symetrycznymi, a w niektórych przypadkach obustronną agenezą płuc. Są również zwykłymi anomaliami układu naczyniowego płuc i dysmorfiami, w tym obustronnym rozszczepem wargi i podniebienia, ankyloglossią, hipoplazją żuchwy, mikroretrognatią i aplazją wargowo-ścięgnistą .
Szenker-Ravi, badając 4 rodziny tetra-Amelii z agenezą lub hipoplazją płuc, zauważył heterogeniczność fenotypową z anomaliami kończyn o różnym nasileniu . Sekwencjonowanie eksomu w tych 4 rodzinach umożliwiło identyfikację obciętych homozygotycznych mutacji w genie RSPO2 . Zespół tetraamelii-2 jest spowodowany homozygotyczną mutacją w genie RSPO2 (610575) zlokalizowanym na chromosomie 8q23 .
fenotyp pierwszego przypadku opisanego w tym artykule odpowiada zespołowi tetra-amelia-1, w szczególności z powodu obecności wodogłowia, anomalii genitaliów i szczątkowego nosa. Wąska klatka piersiowa i wczesna śmierć przed 10 minutą życia sugerują ciężką hipoplazję płuc. W tym przypadku wyróżnia się fenotypową heterogeniczność z kolobomą powieki, hiperteloryzmem, egzoftalmosem i rzadkimi przydatkami.
drugi przypadek w naszym badaniu uważamy za zespół tetramelii-2, biorąc pod uwagę symetryczną tetra-Amelię z obecnością pniaków kończyny górnej. Diagnozę tetra-amelia należy wykonać wcześnie podczas monitorowania ultrasonograficznego. Dlatego należy podnieść świadomość na temat znaczenia monitorowania ultrasonograficznego i wykorzystania 3D / 4D do poprawy wyników badań przesiewowych. Rozpoznanie masy miednicy na USG w połączeniu z Amelią powinno wzbudzić podejrzenie zespołu wady kończyny zespolonej spleno-gonadowej.
ponadto w naszych warunkach należy zachęcać do badania płodu i badania płodu przy użyciu ewoluujących technologii mikromacierzy chromosomowych oraz sekwencjonowania egzomu i genomu. Lepsza charakterystyka przypadków umożliwia udzielanie porad parom i lepszą znajomość tych anomalii klinicznych.
wniosek
zespół Tetra-Amelii jest rzadki i nadal pozostają szare obszary. Te dwa przypadki, w porównaniu z tym, co zostało już opisane w literaturze, ilustrują fenotypową heterogeniczność tetraamelii. Biorąc pod uwagę rzadkie występowanie tych anomalii, ważne byłoby stworzenie międzynarodowego rejestru anomalii w celu zgłaszania przypadków i utworzenia banku próbek do rozszerzonych badań genetycznych dla rodziców.
- Wilcox WR, Coulter CP, Schmitz ML (2015) Clin Perinatol 42: 281-300.
- Bermejo SE, Cuevas L, Amar e (2011) Amelia: wieloośrodkowe opisowe badanie epidemiologiczne w dużym zbiorze danych z Międzynarodowego Clearinghouse for Birth Defects Surveillance and Research oraz przegląd literatury. Am J Med Genet C Semin Med Genet 157: 288-304.
- Zlotogora JSM, Shabany YO, Jarallah ry (1993) Syndrome of tetraamelia with pulmonary hypoplasia. Am J Med Genet 47: 570-571.
- Zimmer EZ (1985) Tetra-amelia z wieloma wadami rozwojowymi u sześciu męskich płodów w jednym krewnym. Europ. J 144: 412-414.
- Rosenak D (1991) Recurrent tetraamelia and pulmonary hypoplasia with multiple malformations in sibs. Am J Med Genet 38: 25-28.
- gershoni BR (1990) Roberts syndrome czy „X-linked amelia”? . Am J Med Genet 37: 569-572.
- Kosaki K, Jones MC, Stayboldt C (1996) Zimmer phocomelia: delineation by principal coordinate analysis. Am J Med Genet 66: 55-59.
- Niemann S(2004) homozygotyczna mutacja WNT3 powoduje tetra-Amelię w dużej spokrewnionej rodzinie. Am J Hum Genet 74: 558-563.
- Krahn m (2005)Tetra-amelia and lung aplasia syndrome: report of a new family and exclusion of candidate genes. Clin Genet 68: 558-560.
- Szenker-RE, Altunoglu U (2018) hamowanie RSPO2 rnf43 i ZNRF3 reguluje rozwój kończyn niezależnie od LGR4 / 5 / 6. Nature 557: 564-569.