okresowe Paraliże
klinicznie użyteczna klasyfikacja pierwotnych okresowych paraliży, przedstawiona w tabeli 1, obejmuje formy hipokalemiczne, hiperkalemiczne i paramiotoniczne.
Tabela 1. Pierwotny paraliż Okresowy (zmodyfikowany z Jurkat-Rott i Lehmann-Horn) (Otwórz tabelę w nowym oknie)
|
choroba |
Gene |
białko |
dziedziczenie |
mutacja |
|
HyperPP |
SCN4A |
Nav1.4 |
Dominujący |
Doskonale |
|
НормоПП |
Mała (ω-pory) |
|||
|
Парамиотония Конгенита |
Doskonale |
|||
|
ГипоПП typu II |
Mała (ω-pory) |
|||
|
ГипоПП Do TEGO, ABY |
CACNA1S |
Кав1.1 |
dominujący |
wzmocnienie (ω-pore) |
|
ThyrotoxicPP |
KCNJ18 |
Kir2.18 |
dominujący |
strata |
|
zespół Andersena-Tawila |
KCNJ2 |
Kir2.1 |
dominujący |
strata |
fizjologiczną podstawą osłabienia wiotkiego jest niewytłumaczalność błony mięśniowej (tj. sarcolemma). Zmiana stężenia potasu w surowicy nie jest główną wadą pierwotnego PP; zmieniony metabolizm potasu jest wynikiem PP. W pierwotnym i tyreotoksycznym PP porażenie wiotkie występuje przy stosunkowo niewielkich zmianach stężenia potasu w surowicy, podczas gdy w wtórnym PP stężenie potasu w surowicy jest znacznie nieprawidłowe.
żaden pojedynczy mechanizm nie jest odpowiedzialny za tę grupę zaburzeń. Są więc niejednorodne, ale mają pewne wspólne cechy. Słabość zwykle jest uogólniona, ale może być zlokalizowana. Zwykle oszczędza się muskulaturę czaszki i mięśnie oddechowe. Odruchy rozciągania są nieobecne lub zmniejszone podczas ataków. Włókna mięśniowe są elektrycznie niewykrywalne podczas ataków. Siła mięśni jest normalna między atakami, ale po kilku latach w niektórych typach PP (zwłaszcza pierwotnego PP) rozwija się pewien stopień stałego osłabienia. Wszystkie formy pierwotnego PP (z wyjątkiem Becker myotonia congenita) są dziedziczone autosomalnie dominująco lub sporadycznie (najprawdopodobniej w wyniku mutacji punktowych).
czułe na napięcie kanały jonowe ściśle regulują generowanie potencjałów działania (krótkie i odwracalne zmiany napięcia błon komórkowych). Są to selektywnie i zmiennie przepuszczalne kanały jonowe. Zależne od energii transportery jonów utrzymują gradienty stężenia. Podczas generowania potencjałów działania jony sodu poruszają się przez membranę przez napięciowe kanały jonowe. Spoczynkowa błona włókien mięśniowych jest spolaryzowana głównie przez ruch chlorku przez kanały chlorkowe i jest repolaryzowana przez ruch potasu. Kanały sodowe, chlorkowe i wapniowe, jako grupa, są związane z miotonią i PP. Funkcjonalne podjednostki kanałów sodowych, wapniowych i potasowych są homologiczne. Kanałopatie sodowe są lepiej poznane niż kanałopatie wapniowe lub chlorkowe. Wszystkie formy rodzinnego PP pokazują końcowy mechanistyczny szlak obejmujący nieprawidłową depolaryzację, inaktywację kanałów sodowych i niewytłumaczalność włókien mięśniowych.
dyskusja w tym artykule dotyczy przede wszystkim kanałów sodowych, wapniowych i potasowych, a także wtórnych form PP. Kanałopatie chlorkowe nie są związane z epizodycznym osłabieniem i są omawiane bardziej szczegółowo w artykułach na temat zaburzeń miotonicznych.
podsumowanie dysfunkcji kanałów w różnych typach PP
przy szybkiej inaktywacji kanału Hiperpp, mutacje są zwykle umiejscowione w wewnętrznych częściach segmentów przezbłonowych lub w pętlach wewnątrzkomórkowych wpływających na miejsca dokowania dla szybko dezaktywującej się cząstki, co upośledza szybką inaktywację kanału prowadzącą do trwałego prądu Na+.
przy hiperpolaryzacji-aktywowanym kationem przeciwdziała prądowi prostującemu K+, mutacje powodują podstawienie argininy lub lizyny.
przy wycieku kationów aktywowanych depolaryzacją NormoPP, mutacje znajdują się w głębszych lokalizacjach czujnika napięcia domeny II w kodonie r675.
dysfunkcja kanału jonowego jest zwykle dobrze kompensowana normalnym wzbudzeniem,A Dodatkowe wyzwalacze są często konieczne do wytworzenia niewytłumaczalności mięśni z powodu długotrwałej depolaryzacji błony.
spożycie glukozy i potasu ma odwrotny skutek w tych zaburzeniach. W Hiperprzebieniu spożycie potasu wywołuje atak, podczas gdy glukoza go łagodzi. W przeciwieństwie do tego, glukoza wywołuje ataki hipokalemiczne, a potas jest leczeniem ataku.
zwróć uwagę na poniższy obrazek.
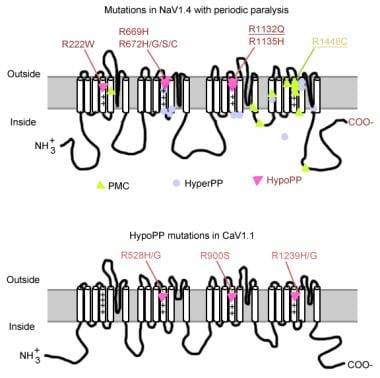 mutacje w paraliżu okresowym.
mutacje w paraliżu okresowym. Gen kanału sodowego mięśni
kanał sodowy ma podjednostkę alfa i podjednostkę beta. Podjednostka Alfa kanału sodowego jest glikoproteiną 260-kd zawierającą około 1800-2000 aminokwasów. Kanał ten jest wysoce zachowany ewolucyjnie od Drosophila do człowieka. Ma 4 domeny homologiczne (I-IV), które składają się, tworząc centralny por, z których każda zawiera 225-325 aminokwasów. Każda domena składa się z 6 hydrofobowych segmentów (S1-S6) przemierzających błonę komórkową. Główne funkcje kanału obejmują bramkowanie wrażliwe na napięcie, inaktywację i selektywność jonów. Zewnątrzkomórkowa pętla między S5 i S6 zanurza się w błonie osocza i bierze udział w tworzeniu porów. Segment S4 zawiera dodatnio naładowane aminokwasy w co trzeciej pozycji i działa jako czujnik napięcia. Podczas depolaryzacji mogą wystąpić zmiany konformacyjne, co prowadzi do aktywacji i dezaktywacji kanału. Pętla komórkowa między domeną III – S6 i domeną IV-S1 działa jako brama inaktywująca.
kanał sodowy ma 2 bramki (aktywację i dezaktywację) i może istnieć w 3 Stanach. W spoczynku, gdy membrana jest spolaryzowana, Brama aktywacyjna jest zamknięta, a Brama inaktywacyjna jest otwarta. W przypadku depolaryzacji Brama aktywacyjna otwiera się, umożliwiając jony sodu przechodzenie przez kanał jonowy, a także odsłaniając miejsce dokowania bramki inaktywacyjnej. Przy ciągłej depolaryzacji Brama inaktywacji zamyka się, blokując wejście sodu do komórki i powodując, że kanał wchodzi w stan szybkiej inaktywacji. Ta inaktywacja kanału umożliwia przepolaryzowanie membrany, co powoduje powrót do stanu spoczynku z zamkniętą bramką aktywacyjną i otwartą bramką inaktywacyjną. W mięśniach szkieletowych ssaków zachodzą dwa procesy inaktywacji: szybka inaktywacja polega na zakończeniu potencjału czynnościowego i działa w skali milisekundowej. Powolna inaktywacja trwa od Sekund Do minut i może regulować populację pobudliwych kanałów sodowych.
mutacje kanału sodowego, które zakłócają szybką i powolną inaktywację, są zwykle związane z fenotypem Hiperpp i miotonii, gdzie jako mutacje, które zwiększają powolną lub szybką inaktywację, powodując utratę funkcji kanału sodowego, powodują Hipopp.
mutacje genu kanału sodowego (SCN4A) mają kilka ogólnych cech. Większość mutacji znajduje się w” inaktywującym „łączniku między powtórzeniami III I IV, w” wykrywającym napięcie ” segmencie S4 powtórzenia IV lub w wewnętrznej błonie, gdzie mogą upośledzić miejsce dokowania dla bramki inaktywacyjnej. Fenotyp kliniczny różni się specyficzną substytucją aminokwasów i, chociaż niektóre nakładanie się może wystąpić między hiperkalemicznym PP, paramiotonia congenita (PC) i miotoniami obciążonymi potasem (PAM), 3 fenotypy są ogólnie różne (jak opisano poniżej). Prawie wszystkie zmutowane kanały mają zaburzenia szybkiej inaktywacji prądu sodowego. Większość pacjentów jest wrażliwa na potas ogólnoustrojowy lub na niską temperaturę.
istnieją dwie populacje kanałów, zmutowane i typu dzikiego; zaburzona szybka inaktywacja powoduje przedłużoną depolaryzację zmutowanych błon włókien mięśniowych i może wyjaśnić 2 główne objawy tych zaburzeń, miotonię i osłabienie. W hiperkalemicznym PP następuje wzmocnienie funkcji w zmutowanym bramkowaniu kanału, co skutkuje zwiększonym prądem sodowym nadmiernie depolaryzującym dotknięty mięsień. Łagodna depolaryzacja (5-10 mV) błony mięśniowej, która może być spowodowana zwiększonym zewnątrzkomórkowym stężeniem potasu, powoduje, że zmutowane kanały są utrzymywane w trybie nieinaktywowanym. Uporczywy Wewnętrzny prąd sodowy powoduje powtarzające się wypalanie kanałów sodowych typu dzikiego, co jest postrzegane jako sztywność (tj. miotonia).
jeśli występuje poważniejsza depolaryzacja (20-30 mV), zarówno normalne, jak i nieprawidłowe kanały są ustalone w stanie inaktywacji, powodując osłabienie lub paraliż. Tak więc subtelne różnice w nasileniu depolaryzacji błon mogą odróżniać miotonię od paraliżu. Czułość temperaturowa jest cechą charakterystyczną komputera. Zimno zaostrza miotonię i wywołuje osłabienie. Z tą chorobą wiąże się szereg mutacji, z czego 3 w tym samym miejscu (1448) w segmencie S4. Mutacje te zastępują argininę innymi aminokwasami i neutralizują ten wysoce zachowany ładunek dodatni S4. Mutacje tych pozostałości są najczęstszą przyczyną PC. Niektóre z możliwych mechanizmów odpowiedzialnych za czułość temperaturową obejmują następujące:
-
temperatura może w różny sposób wpływać na zmianę konformacyjną kanału mutanta.
-
niższe temperatury mogą ustabilizować kanały mutantów w nienormalnym stanie.
-
mutacje mogą zmieniać wrażliwość kanału na inne procesy komórkowe, takie jak fosforylacja lub wtórni posłańcy.
większość przypadków hiperkalemicznego PP wynika z 2 mutacji w SCN4A, T704M i M1592V. Mutacje w kanale sodowym, zwłaszcza w resztach 1448 i 1313, są odpowiedzialne za paramyotonia congenita. Niewielki odsetek przypadków hipokalemicznego paraliżu okresowego wiąże się z mutacjami w kodonach 669 i 672 (HypoPP2). W Hipopp2 mutacje kanału sodowego zwiększają inaktywację w celu wytworzenia utraty funkcji netto.
Normokalemiczny PP przypomina zarówno HyperPP (czułość potasu), jak i HypoPP (czas trwania ataków) i jest spowodowany mutacjami SCN4A w głębszej lokalizacji czujnika napięcia DII w kodonie 675. Mutacje R675 różnią się od Hipopp tym, że mutacje te powodują aktywowane przez depolaryzację otwory bramkowe generujące prąd ω z odwróconą zależnością napięciową, ponieważ miejsce to jest narażone na miejsca zewnątrzkomórkowe przy silniejszej depolaryzacji.
Gen kanału wapniowego
Gen kanału wapniowego (CACNL1A3) jest kompleksem 5 podjednostek (alfa-1, alfa-2, beta, gamma i delta). Receptor dihydropirydyny mięśni szkieletowych (DHP) znajduje się głównie w poprzecznej błonie kanalikowej. Podjednostka alfa-1 ma miejsca wiążące dla leków DHP i prowadzi powolny prąd wapniowy typu L. Bierze również udział w sprzężeniu wzbudzenie-skurcz (EC) i działa jako czujnik napięcia poprzez połączenie z receptorem ryanodine retikulum sarkoplazmatycznego (tj. kanał uwalniania wapnia). Wszelkie zmiany potencjału błonowego są związane z wewnątrzkomórkowym uwalnianiem wapnia, co umożliwia sprzęganie EC. Mutacje punktowe w podjednostce receptora DHP/kanału wapniowego alfa-1 powodują hipokaliemię PP (HypoPP1). Dwie mutacje genu CACNA1S, R528H i R1239H, są odpowiedzialne za większość przypadków HIPOKALEMICZNEGO PP.
fizjologiczna podstawa choroby nadal nie jest zrozumiała, ale jest bardziej prawdopodobna z powodu awarii wzbudzenia, niż awarii sprzężenia we. Jednak depolaryzacja wywołana hipokalemią może zmniejszyć uwalnianie wapnia, wpływając bezpośrednio lub pośrednio na kontrolę napięcia kanału poprzez inaktywację kanału sodowego. Insulina i adrenalina mogą działać w podobny sposób. Mutacje genu kanału wapniowego mają pewne podobieństwa do mutacji SCN4A. Mutacje modyfikują inaktywację kanałów, ale nie aktywację zależną od napięcia. Nagrania z hodowli myotube od chorych pacjentów wykazały 30% redukcję czułego na DHP prądu wapniowego typu L. Kanały są inaktywowane przy niskim potencjale membranowym.
mutacje kanału wapniowego powodują utratę funkcji objawiającą się zmniejszoną gęstością prądu i wolniejszą inaktywacją. Nie wiadomo, w jaki sposób ta inaktywacja jest związana z atakami wywołanymi hipokalemią. Co najmniej w mutacji R528H występuje możliwa wtórna kanałopatia związana ze zmniejszeniem wrażliwego na ATP prądu potasowego ze zmienionej homeostazy wapnia. Niższe prądy związane z mutacjami CACNL1A3 mogą nieznacznie zmienić wewnątrzkomórkową homeostazę wapnia, co może wpływać na właściwości i ekspresję kanałów K+, w szczególności kanału potasowego katp (ATP-sensitive potas channel) należącego do klasy kanałów prostujących wewnątrzkomórkowych. Insulina działa również w HypoPP zmniejszając ten wewnętrzny prostownik K + prąd.
utrata ładunku czujnika napięcia w większości przypadków Hipopp. Kanały sodowe i wapniowe mają homologiczne podjednostki alfa tworzące pory. Mutacje punktowe w CACNL1A3 i SCN4A wpływają na pozostałości w czujnikach napięcia S4 tych kanałów. Mutacje argininy w segmentach S4 są odpowiedzialne za 90% przypadków Hipopp.
utrata ładunku czujnika napięcia w większości przypadków Hipopp. Kanały sodowe i wapniowe mają homologiczne podjednostki α tworzące pory. Prawie wszystkie mutacje Cav1.1 (HypoPP-1) i Nav1.4 (HypoPP-2) neutralizują dodatnio naładowany aminokwas w jednej z najbardziej oddalonych arginin lub lizyn czujników napięcia. Nav1.4 mutacje znajdują się najczęściej w czujnikach napięcia powtórzeń I, II i III, powodując wyciek kationów.
Podstawienie najbardziej zewnętrznej argininy mniejszym aminokwasem, takim jak glicyna, otwiera szlak przewodzący przy hiperpolaryzowanym potencjale, co skutkuje wewnętrznym prądem kationowym (wyciek kationowy lub prąd ω w celu odróżnienia od (ω-) przez porów przewodzących jony, jest aktywowanym przez hiperpolaryzację prądem monowalentnych kationów poprzez S4 bramkowanie porów przeciwdziałającym prostowaniu prądów K+) depolaryzującym lub destabilizującym potencjał spoczynkowy.
segment S4 porusza się na zewnątrz podczas depolaryzacji zamykając szlak przewodzący. Włókna mięśniowe z silnymi mutacjami czujnika napięcia ulegają depolaryzacji nie tylko podczas hipokaliemii, ale także na poziomie potasu w normie, co tłumaczy wewnętrzne i trwałe osłabienie. Ciężka miopatia z tłuszczowym zastąpieniem tkanki mięśniowej jest powszechnie stwierdzana u pacjentów z mutacjami Cav1.1 R1239h (div).
glikokortykosteroidy powodują Hipopp poprzez stymulację Na + K + ATPazy za pośrednictwem insuliny i amyliny.
Gen kanału potasowego
wewnętrzna rektyfikacja jest ważną właściwością kanałów Kir. Rektyfikacja polega na zależnym od napięcia przewodzeniu-zatkaniu porów poliaminami i Mg++ podczas depolaryzacji, a zatkanie to jest usuwane podczas gradientu potencjalnego podczas hiperpolaryzacji. Mutacje kanału potasowego obserwuje się w zespole Andersena-Tawila i tyreotoksycznym PP.
Triada cech dysmorficznych, okresowe porażenie i zaburzenia rytmu serca charakteryzuje zespół Andersena-Tawila. Zespół ten jest związany z mutacjami w genie KCNJ2. Gen KCNJ2 koduje wewnętrzny kanał potasowy Kir2.1. Mutacje kanału potasowego w KCNE3 powodują hipokaliemię PP, ale nie zostało to potwierdzone.
mutacje w Kir2. 6 powodują wrażliwość na tyreotoksyczność PP. Epizodyczne osłabienie obserwowane w tyreotoksycznym PP jest podobne do obserwowanego w hipoppie i zespole Andersena-Taville ’ a. To zaburzenie jest najbardziej rozpowszechnione u Azjatów i latynoamerykańskich mężczyzn. Tyreotoksyczny PP jest chorobą genetyczną zdemaskowaną przez tyreotoksykozę. Kir2. 6 wyraża się głównie w mięśniach szkieletowych. Trijodotyronina wzmacnia transkrypcję KCNJ18, co może prowadzić do zwiększonej ekspresji Kir2. 6. PKC jest aktywowany podczas tyreotoksykozy z powodu zwiększonego obrotu PIP2 i kanałów Kir bezpośrednio oddziałują z PIP2 podczas normalnego bramkowania. W zespole Andersena-Tawila występuje zmniejszone powinowactwo PIP2. W tyreotoksycznym PP żadna z mutacji nie zmienia rektyfikacji Kir2. 6.