Zespół aicardiego-Goutières 'a: spektrum fenotypowe i genetyczne w serii trzech przypadków | Anales de Pediatría
zespół AICARDIEGO-Goutières’ a (AGS) jest rzadką chorobą dziedziczną, której częstość występowania jest nieznana. Po raz pierwszy został opisany w 1984 roku przez Jeana Aicardiego i Francoise Goutières jako postępująca encefalopatia z początkiem w pierwszych miesiącach życia charakteryzująca się limfocytozą płynu mózgowo-rdzeniowego (CSF) i zwapnieniami w zwojach podstawnych.1 objawia się drażliwością, opóźnieniem psychoruchowym, spastycznością, dystonią, napadami padaczkowymi, nawracającymi epizodami aseptycznej gorączki i małogłowiem. Śmiertelność jest wyższa w fazie encefalopatycznej i chociaż choroba zwykle stabilizuje się później, powoduje ciężkie następstwa neurologiczne. Inne charakterystyczne cechy, które mogą pojawić się w trakcie jego przebiegu, to chilblains, objawy oczne (głównie jaskra), zajęcie serca lub choroby autoimmunologiczne.2 interferony typu i odgrywają kluczową rolę w patogenezie AGS, w której ich ekspresja jest regulowana, co prowadzi do zwiększenia produkcji.Z tego powodu jednym z klasycznych wyników badań laboratoryjnych u tych pacjentów jest podwyższony poziom interferonu alfa w płynie mózgowo-rdzeniowym, wraz z pleocytozą i równie podwyższonym poziomem neopteryny i biopteryny. Potencjalna przydatność oceny poziomu ekspresji genów stymulowanych interferonem przez interferon we krwi obwodowej jako markera jest obecnie badana, ponieważ istnieją dowody na to, że poziomy te pozostają wysokie po fazie encefalopatycznej („interferon signature”).3-5 inną kluczową cechą jest wykrywanie nieprawidłowości neuroobrazowania, w tym zwapnień w zwojach podstawnych i zmian w istocie białej (Fig. 1). Do tej pory znamy 7 genów, których mutacje mogą prowadzić do zwiększenia regulacji szlaku interferonu: ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1 i IFIH1. Heterozygotyczne mutacje zostały opisane dla genów TREX1, ADAR I IFIH1, podczas gdy mutacje zgłaszane we wszystkich innych genach były homozygotyczne.2 mutacje w genie IFIH1 wykryto ostatnio (2014 r.) 4 i dlatego są najmniej częstymi wariantami chorobotwórczymi, podczas gdy mutacje w genach RNASEH2B i TREX1 stanowią najwyższy odsetek zdiagnozowanych przypadków AGS.
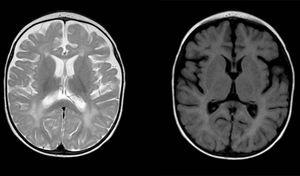
rozproszone i niejednoznaczne nieprawidłowości sygnałowe w istocie białej na obu półkuli mózgowej, hiperintensywność w obrazach ważonych T2. Powiększona przestrzeń podpajęczynówkowa z przewagą przednio-skroniową w obu półkulach, z poszerzeniem szczeliny między półkulowej i zwiększeniem wielkości komór (przy braku zwiększonego ciśnienia), zgodna z zanikiem korowym i podkorowym.
w ciągu ostatnich kilku dekad, dzięki postępowi w genetyce umożliwiającemu wykrywanie tych specyficznych mutacji, pojawiły się dowody na szerokie spektrum fenotypowe poza klasyczną prezentacją opartą na Genie sprawczym. Przedstawiamy przypadki 3 pacjentów, którym w ciągu ostatnich 8 lat postawiono diagnozę AGS w celu analizy ich cech klinicznych w odniesieniu do podstawowej wady genetycznej (Tabela 1). Ogólnie rzecz biorąc, przedstawione cechy AGS były zgodne z opisanymi w najnowszej serii przypadków w literaturze: prezentacja noworodka (33%), małogłowie (66%), opóźnienie psychoruchowe (100%), spastyczność (100%), ciężka niepełnosprawność intelektualna (66%) i zwapnienia na tomografii komputerowej czaszki (66%), chociaż tylko jeden pacjent miał napady padaczkowe.
charakterystyka pacjentów z zespołem Aicardi-Goutières.
| Przypadek 1 | Przypadek 2 | Przypadek 3 | |
|---|---|---|---|
| genetyka | mutacja homozygotyczna (P. Ala177Thr) w genie RNASEH2B | mutacja homozygotyczna (341G>A) w genie TREX1 | mutacja Heterozygotyczna (c.992C>G I p.Thr331Arg) w genie IFIH1 |
| obecny wiek | 3 lata | 7 lat i 4 miesiące | 12 lat i 11 miesięcy |
| płeć | Mężczyzna | Kobieta | Mężczyzna |
| pochodzenie | Rumunia | Hiszpania | Włochy |
| AP | – | Tydzień 36: wewnątrzmaciczne ograniczenie wzrostu Tydzień 37: małogłowie, zwapnienie łożyska |
rozszczep podniebienia |
| objawy kliniczne | |||
| Wiek na początku | 10 miesięcy | poród | 2 lata |
| Prezentacja początkowa | drażliwość regresja Psychomotoryczna |
drżenia, hipotonia, słaby płacz, niewydolność wzrostu | opóźnienie zdolności motorycznych |
| upośledzenie psychoruchowe | tak | tak | tak |
| język | 2-Sylabowy words | No | Yes |
| Microcephaly | Yes | Yes | No |
| Chilblains | No | Yes | Yes |
| Epilepsy | No | Yes | No |
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFC IV |
| zaburzenia ruchu | Nie | tak | Nie |
| nieprawidłowe ruchy gałek ocznych | Nie | Nie | Nie |
| zaburzenia widzenia | Nie | – | krótkowzroczność |
| jaskra | Nie | Nie | Nie |
| utrata słuchu | – | – | Nie. |
| zaburzenia czynności serca | Nie | łagodna niedomykalność trójdzielna i mitralna | Nie |
| nawracająca gorączka | Nie | Nie | Nie |
| niepełnosprawność intelektualna | tak | tak, ciężki | tak, łagodny |
| Inne | – | – | zespół Singletona-Mertena: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
| CSF | |||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – |
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – |
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – |
| INF alfa (IU/mL) | 25 ( | 75 ( | – |
| Blood | |||
| Interferon signature | – | – | Gene overexpression |
| Neuroimaging | |||
| Calcifications on CT | No | Yes. | tak . Symetryczne zwapnienia w głębokim WM okolicy czołowej i jądra soczewicy |
| MRI głowy | rozproszone i niejednoznaczne zmiany intensywności WM obu półkul mózgowych, hiperintense na T2. Udział podkorowych włókien WM (oszczędzających włókna U) i okołoporodowych WM | uogólniony udział WM z przewagą płata wm, w tym podkorowych włókien u płata czołowego, skroniowego i potylicznego, obustronnie i symetrycznie, bez udziału korowego | – |
CSF, płyn mózgowo-rdzeniowy; CT, tomografia komputerowa; GMFC, System Klasyfikacji funkcji motorycznych brutto; INF, interferon; IUGR, ograniczenie wzrostu wewnątrzmacicznego; MRI, rezonans magnetyczny; PNP, polineuropatia; WM, Biała mater.
jak wspomniano wcześniej, homozygotyczne mutacje w genie RNASEH2B są najczęstszymi wariantami powodującymi AGS, a ich ekspresja fenotypowa zwykle najbardziej odpowiada klasycznej prezentacji.4 tak było w przypadku pacjenta w naszym badaniu, który posiadał taką mutację, który miał początek w wieku 10 miesięcy z drażliwością i opóźnieniem psychomotorycznym oraz z charakterystycznymi objawami neuroobrazowania i CSF.
dwadzieścia procent przypadków AGS może mieć postać noworodka, z początkiem choroby występującej w macicy.5 mutacje w którymkolwiek z 7 wyżej wymienionych genów mogą prowadzić do tego fenotypu, ale ta wczesna prezentacja jest najczęściej związana z genem TREX.Początkowa postać tej postaci jest podobna do postaci zakażenia TORCH, z powiększeniem wątroby, hipertransaminazemią, małopłytkowością i objawami neurologicznymi, w tym skrajną drażliwością, zaburzeniami ruchowymi i napadami padaczkowymi.Pacjenci ci mają cięższy przebieg choroby i są narażeni na większe ryzyko zgonu. Pacjent w naszej próbce, który przedstawił taki wariant, miał prezentację noworodkową i obecnie ma najcięższą postać choroby z 3.
mutacje w genie ADAR1, a zwłaszcza w genie IFIH1 są związane z późnym początkiem objawów, po 1 roku życia z prawidłowym rozwojem psychomotorycznym.5 w niektórych z tych przypadków zespół ma łagodny przebieg ze względnym zachowaniem umiejętności językowych i motorycznych. Nasz pacjent z mutacją w genie IFIH1 był pojedynczym przypadkiem, ponieważ miał również zespół Singletona-Mertena, rzadką chorobę również spowodowaną mutacją w genie IFIH1, charakteryzującą się dysplazją zębów, zwapnieniami aorty i osteoporozą.6
naszym celem jest podkreślenie istotnej zmienności fenotypowej AGS i jej związku z konkretnymi mutacjami w celu zarówno zachęcenia do rozważenia tej diagnozy w przypadkach, w których prezentacje odbiegają od klasycznej postaci choroby, jak i dostarczenia dodatkowych informacji na temat przebiegu choroby i wyników u tych pacjentów.