Aicardi-Goutières síndrome: Fenotípica e genética do espectro em uma série de três casos | Anales de Pediatría
Aicardi-Goutières síndrome (AAC) é uma doença hereditária rara, cuja prevalência exacta é desconhecida. Foi descrita pela primeira vez em 1984 por Jean Aicardi e Françoise Goutières como uma encefalopatia progressiva, com início nos primeiros meses de vida, caracterizado por líquido cefalorraquidiano (LCR) linfocitose e calcificações nos gânglios da base.Manifesta-se com irritabilidade, atraso psicomotor, espasticidade, distonia, crises epilépticas, episódios recorrentes de febre asséptica e microcefalia. A mortalidade é maior durante a fase encefalopática e, embora a doença tipicamente estabilize depois, causa sequelas neurológicas graves. Outras características que podem aparecer durante o seu curso são chilblains, sintomas oculares (principalmente glaucoma), envolvimento cardíaco ou distúrbios auto-imunes.2 os interferões tipo I desempenham um papel crucial na patogênese da AGS, na qual a sua expressão é re-regulada levando ao aumento da produção.Por esta razão, um dos resultados laboratoriais clássicos nestes doentes é um nível elevado de interferão alfa no líquido cefalorraquidiano, juntamente com pleocitose e níveis igualmente elevados de neopterina e biopterina. A potencial utilidade de avaliar o nível de expressão dos genes estimulados pelo interferão no sangue periférico como marcador está actualmente a ser investigada, uma vez que existem provas de que estes níveis se mantêm elevados para além da fase encefalopática (“assinatura do interferão”).3-5 outra característica chave é a detecção de anormalidades de neuroimagem, incluindo calcificações nos gânglios basais e mudanças na matéria branca (Fig. 1). Até à data, conhecemos 7 genes cujas mutações podem levar à regulação da Via do interferão: ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1 e IFIH1. As mutações heterozigóticas foram descritas para os genes TREX1, ADAR e IFIH1, enquanto as mutações notificadas em todos os outros genes foram homozigóticas.2 mutações no gene IFIH1 foram detectadas mais recentemente (2014)4 e são, portanto, as variantes patogénicas menos frequentes, enquanto as mutações nos genes RNASEH2B e TREX1 representam a maior proporção de casos diagnosticados de AGS.
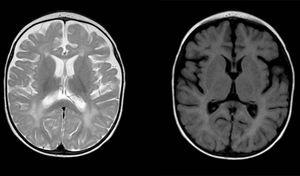
anomalias difusas e irregulares do sinal na matéria branca em ambos os hemisférios cerebrais, hiperintensa em imagens ponderadas em T2. Espaço subaracnóide alargado com predominância frontotemporal em ambos os hemisférios, com alargamento da fissura interemisférica e aumento do tamanho ventricular (na ausência de aumento de pressão), compatível com atrofia cortical e subcortical.
Nas últimas décadas, graças aos avanços em genética, permitindo a detecção destas mutações pontuais, a evidência surgiu de uma ampla fenotípica espectro além da clássica apresentação com base no gene causal. Apresentamos os casos de 3 pacientes que receberam um diagnóstico de AGS nos últimos 8 anos com o objetivo de analisar suas características clínicas em relação ao defeito genético subjacente (Tabela 1). Em geral, a apresentar características de AGS foram consistentes com aqueles descritos na mais recente série de casos na literatura: apresentação neonatal (33%), microcefalia (66%), retardo do desenvolvimento psicomotor (100%), espasticidade (100%), severa deficiência intelectual (66%) e calcificações no craniana CT (66%), embora apenas um paciente teve crises epilépticas.
características dos doentes com síndrome de Aicardi-Gotières.
| Caso 1 | Caso 2 | Caso 3 | |
|---|---|---|---|
| Genética | mutação Homozigótica (p.Ala177Thr) em RNASEH2B gene | mutação Homozigótica (341 G>A) em TREX1 gene | mutação Heterozigótica (c.992C>G e p.Thr331Arg) no gene IFIH1 |
| idade Atual | 3 anos | 7 anos e 4 meses | 12 anos e 11 meses |
| Sexo | Masculino | Feminino | Macho |
| Origem | Roménia | Espanha | Itália |
| AP | – | Semana 36: restrição de crescimento intra-uterino Semana 37: microcefalia, calcificação placentária |
Fenda palatina |
| manifestações Clínicas | |||
| a Idade no início | 10 meses | Nascimento | 2 anos |
| Apresentação inicial | Irritabilidade regressão Psicomotora |
Tremores, hipotonia, choro fraco, dificuldade de crescimento | habilidade Motora atraso |
| retardo do desenvolvimento Psicomotor | Sim | Sim | Sim |
| Idioma | 2-Sílaba words | No | Yes |
| Microcephaly | Yes | Yes | No |
| Chilblains | No | Yes | Yes |
| Epilepsy | No | Yes | No |
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFCS IV |
| distúrbio de Movimento | Não | Sim | Não |
| Anormal dos movimentos oculares | Não | Não | Não |
| deficiência Visual | Não | – | Miopia |
| Glaucoma | Não | Não | Não |
| perda de Audição | – | – | Nenhum |
| Envolvimento cardíaco | Não | Leve mitral e regurgitação mitral | Não |
| febre Recorrente | Não | Não | Não |
| deficiência Intelectual | Sim | Sim, grave | Sim, leve |
| Outros | – | – | Singleton-Merten síndrome de: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
| CSF | |||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – |
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – |
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – |
| INF alfa (IU/mL) | 25 ( | 75 ( | – |
| Blood | |||
| Interferon signature | – | – | Gene overexpression |
| Neuroimaging | |||
| Calcifications on CT | No | Yes. Em gânglios basais e periventriculares | Sim. Calcificações simétricas em WM profundo da região frontal e do núcleo dentiforme |
| IRM cabeça | alterações difusas e estaladiças na intensidade WM de ambos os hemisférios cerebrais, hiperintensa no T2. O envolvimento de subcortical WM (poupando o U fibras) e periventricular WM | Generalizada WM envolvimento com predominância de lobar WM incluindo subcorticais U fibras do frontal, temporal e occipital lobos, a nível bilateral e simetricamente, sem envolvimento cortical | – |
LCR, líquido cefalorraquidiano; CT, tomografia computadorizada; GMFCS, Gross Motor Function Classification System; INF, interferon; IUGR, restrição de crescimento intra-uterino; ressonância magnética ressonância magnética; PNP, polineuropatia; WM, branco mater.
como observado antes, as mutações homozigóticas no gene RNASEH2B são as variantes mais frequentes que causam AGS e sua expressão fenotípica geralmente se conforma com a apresentação clássica.Este foi o caso do paciente em nosso estudo que realizou tal mutação, que teve início aos 10 meses de idade com irritabilidade e atraso psicomotor e com características de neuroimagem e resultados CSF.
vinte por cento dos casos de AGS podem ter uma apresentação neonatal, com o início da doença ocorrendo no útero.5 mutações em qualquer um dos 7 genes acima mencionados podem levar a este fenótipo, mas esta apresentação precoce é mais frequentemente associada com o gene TREX.4,5 a apresentação inicial desta forma é semelhante à de uma infecção por tocha, com hepatosplenomegalia, hipertransaminasemia, trombocitopenia e manifestações neurológicas incluindo irritabilidade extrema, distúrbios do movimento e convulsões epilépticas.Estes pacientes têm um curso mais grave de doença e estão em maior risco de morte. O paciente em nossa amostra que apresentou tal variante teve uma apresentação neonatal e atualmente tem a forma mais grave de doença do 3.
as mutações no gene ADAR1 e especialmente no gene IFIH1 estão associadas a um início tardio dos sintomas, após 1 ano de vida com o desenvolvimento psicomotor normal.5 em alguns destes casos, a síndrome tem um curso benigno com relativa preservação da linguagem e habilidades motoras. O nosso paciente com uma mutação no gene IFIH1 foi um caso singular na medida em que também tinha síndrome de Singleton-Merten, uma doença rara também causada por uma mutação no gene IFIH1 e caracterizada por displasia dentária, calcificações aórticas e osteoporose.6
o Nosso objetivo é destacar a significativa variabilidade fenotípica de AGS e sua associação com mutações pontuais a fim de incentivar a considerar este diagnóstico em casos de apresentações que se afastam da forma clássica da doença e de contribuir com informações adicionais sobre o curso da doença e os resultados desses pacientes.