Gynecology & Obstetrics Case report
Keywords
Tetra-amelia; malformação; genótipo; fenótipo.
introdução
as anomalias dos Membros constituem um grupo importante de patologias congénitas caracterizadas por hipoplasia ou aplasia completa de um ou mais ossos dos membros. Anomalias dos membros de todos os tipos ocorrem em aproximadamente 1 em 1.300 a 2.000 nascimentos. Estas anomalias nos membros podem ser isoladas ou associadas a outras anomalias . A síndrome de Tetraamelia é rara e as áreas cinzentas permanecem.
relatamos dois casos de tetra-amelia, em um nível II de maternidade em Dakar (Senegal) para ser semelhante a tetraamelia-1 (cromossomo 17q21), tetraamelia-2 (cromossomo 8q23) e Robert (síndrome do cromossomo 8p21). Isto ilustra a dificuldade em correlacionar fenótipo e genes envolvidos.
relatórios de casos
Caso 1
a Sra. AD foi uma mãe de 44 anos de idade encaminhada para o nosso departamento às 36 semanas de gestação com graves pré-eclampsia e anomalias fetais. Ela era cinco para sem histórico de anomalias fetais. Ela não fumava agora e nunca tinha fumado nem bebido álcool. Ela não tinha sido exposta ao fumo passivo. Ela estava em um casamento consanguíneo de terceiro grau para todos os seus filhos. A Sra. AD deu negativo para hepatite B, VIH e sífilis. Ela foi protegida do vírus da rubéola e não teve exposição prévia a Toxoplasma gondii. A monitorização ultrassom realizada no final das 33 semanas e 35 semanas de gestação recuperou oligoamniose e hidrocefalia, bem como agenese dos membros. As prescrições durante a gravidez incluíram a administração de ferro e ácido fólico, bem como a administração de sulfadoxina pirimetamina. Este último foi prescrito às 18 semanas e depois às 26 semanas como parte da Política de profilaxia anti-malária para mulheres grávidas. A altura da sínfise-Funda era de 28 cm. Devido a características graves da pré-eclampsia, ela foi hospitalizada imediatamente e observada em uma unidade de parto e parto. Ela então recebeu inicialmente sulfato de magnésio IV para prevenir eclampsia e medicamentos anti-hipertensores para manter a pressão arterial sistólica abaixo de 160 mmHg e pressão arterial diastólica abaixo de 105 mmHg.
a decisão de entrega imediata cesariana foi tomada. Um vivo de 2,150 gramas foi extraído que posteriormente morreu em 10 minutos. Antes da morte, o corpo apresentava movimentos rastejantes. Foram identificadas várias anomalias externas (Figura 1), incluindo agenese completa dos quatro membros, hidrocefalia com uma circunferência da cabeça de 39 cm. No rosto, havia hipertelorismo com um coloboma das pálpebras, exoftalmos leves e aniridia. A boca era semelhante a um V invertido sem clara delimitação dos lábios e o nariz era rudimentar. O recém-nascido era desprovido de integumentos (cabelo e sobrancelhas). As orelhas foram reduzidas a esboços que pareciam fendas. O pescoço era curto. O tronco foi reduzido a uma estrutura cónica de 26 cm de comprimento com um cordão umbilical na extremidade inferior. O peito era estreito. Logo abaixo do umbigo, o tronco estendido para trás estava presente no chão do Polo caudal, uma linha média com recessões e uma brotação que pode corresponder a um falo de tipo indeterminado. Observou-se agenese da pélvis, genitais e imperforação anal. A patologia Fetal não foi realizada. No entanto, a morte dentro de 10 minutos após o parto e a aparência cónica do tórax pode sugerir anomalias pulmonares.
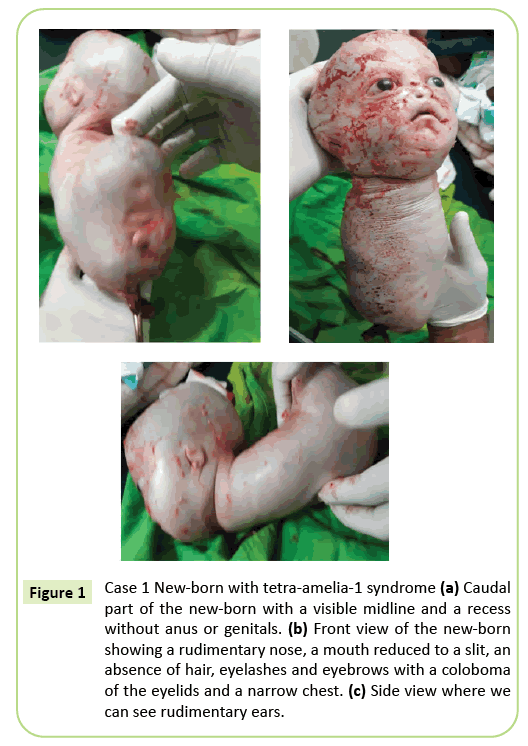
Figura 1: Caso 1 recém-nascido com síndrome de tetra-amelia-1 (a) parte Caudal do recém-nascido com uma linha média visível e um recesso sem ânus ou genitais. b) Visão Frontal do recém-nascido com um nariz rudimentar, uma boca reduzida a uma fenda, ausência de cabelo, pestanas e sobrancelhas com um coloboma das pálpebras e um peito estreito. (C) visão lateral onde podemos ver orelhas rudimentares.
caso 2
o segundo caso foi um primigravida de 22 anos de idade, referido à nossa instalação para ecografia a 37 semanas de gestação. Ela não estava num casamento consanguíneo. Deu negativo para hepatite B, HIV e sífilis. Ela não foi testada para toxoplasmose e rubéola. Não foi feita nenhuma ecografia durante a gravidez. O exame clínico foi consistente com atraso no crescimento fetal (altura Fundal: 26 cm). Os resultados da ecografia mostraram que o úmero estava distorcido, medindo 23,9 mm, correspondendo a 17 semanas de gestação. Houve agenese do fémur. As asas ilíacas eram visíveis no ultra-som. Não foram identificadas anomalias pulmonares ou cardíacas. A entrega foi iniciada. O recém-nascido tinha um fenótipo feminino com uma pontuação Apgar de 9 aos 5 minutos. A morfologia da cabeça e do tronco era sem particularidade. Os membros superiores foram reduzidos para dois tocos de 3 cm de comprimento. Foi observada agenese completa dos 2 membros inferiores. Foi uma anomalia simétrica (Figura 2).
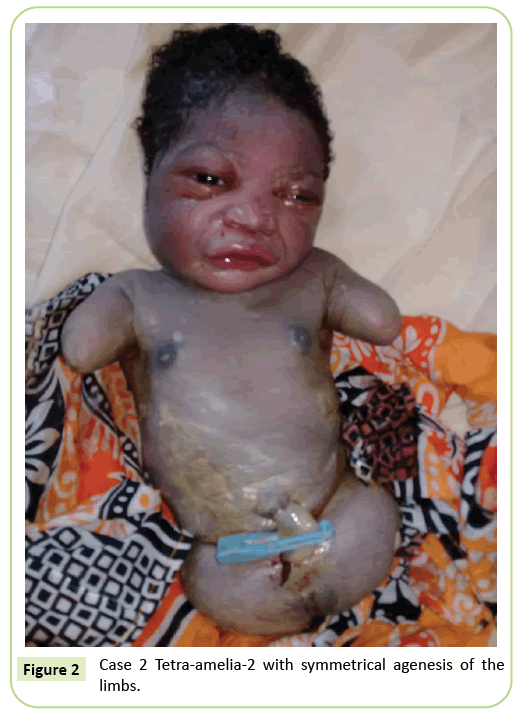
Figura 2: Caso 2 Tetra-amelia-2 simétricas e agenesia de membros.
Discussão
Bermejo-Sanchez et al. descrito em 2011 a epidemiologia da Amelia congênita usando dados coletados de 20 programas de vigilância de anomalias congênitas, de todos os continentes, exceto África, entre 1968 e 2006. No total, 326 casos de Amélia foram identificados entre 23.110.591 nados – vivos, nados-mortos e abortos. A prevalência foi de 1,41 / 100,000 .
Tetra-amelia refere-se à ausência completa dos Membros e ocorre mais raramente. Tanto quanto sabemos, a tetra-amelia-1 é descrita em 7 famílias. Parece seguir uma herança recessiva autossómica. Em todas as famílias, a tetra-amelia – 1 estava associada a malformações graves das outras partes do corpo, incluindo a face e a cabeça, anomalias do sistema nervoso, esqueleto e genitália. Os pulmões estavam subdesenvolvidos em muitos casos, o que torna a respiração difícil ou impossível . Zimmer et al. relatou em 1985 uma família fortemente incrustada na qual 6 crianças tiveram tetra-amelia-1 e hidrocefalia. Eles descreveram em um dos fetos uma ausência total de osso pélvico, lábio leporino e palato, arrínia e aplasia das orelhas. Um pulmão esquerdo bilateral, um canal arterial persistente, uma imperforação anal também foram observados. O teste Fetal eliminou o diagnóstico da síndrome de Robert . Outros casos encontrados na literatura incluem o de Kosaki et al., em 1996, com um feto do cariótipo 46, XX com tetrafocomelia e hipoplasia pulmonar grave, além de anomalias da face e da cabeça . Rosenak et al. descreveu um caso de tetra-amelia com hipoplasia pulmonar grave em dois fetos de um casal não consanguíneo. O teste Fetal excluiu o diagnóstico da síndrome de Robert . Foram notificados dois casos adicionais por Zlotogora et al. em 1993. Ambos os pacientes morreram logo após o nascimento e os autores sugeriram a existência de hipoplasia pulmonar. Niemann et al. relatou uma família consanguínea turca na qual 4 dos 8 irmãos sofriam de tetra-Amélia. Além da ausência dos 4 membros, os exames fetais de 3 fetos revelaram múltiplas anomalias: lábios fissurados e /ou Palatino, laparoscis, anomalias pulmonares, hipoplasia da pélvis, atresia das choanas, vagina e imperforação anal . Finalmente, em 2005, Krahn et al. descreveu dois irmãos nascidos de pais inatos que sofriam de tetraamelia e hipoplasia pulmonar grave. As clavículas e omoplatas estavam normais no segundo feto. O cariótipo era normal .
síndrome Tetra-amelia-1 ou TETAMS1 é causada por uma mutação homozigótica no gene WNT3 no cromossoma 17q21 com uma herança recessiva autossómica. A síndrome Tetraamelia-2 (TETAMS2) é caracterizada por membros rudimentares ou uma ausência completa dos membros, geralmente simétrica, bem como agenese bilateral dos pulmões, em alguns casos. São também anomalias usuais do sistema vascular pulmonar e dismorfias, incluindo fissura bilateral no lábio e palato, anquiloglossia, hipoplasia mandibular, microretrognatia e aplasia labioscrotal .
Szenker-Ravi, estudando 4 famílias de tetra-amelia, com agenesia ou hipoplasia pulmonar, observou uma heterogeneidade fenotípica com o membro anomalias de gravidade variável . A sequenciação de Exome nestas 4 famílias tornou possível identificar mutações homozigóticas truncantes no gene RSPO2 . A síndrome Tetraamelia-2 é causada por uma mutação homozigótica no gene RSPO2 (610575) localizado no cromossoma 8q23 .
o fenótipo do primeiro caso descrito neste artigo corresponde a uma síndrome tetra-amelia-1 devido, em particular, à presença de hidrocefalia, anomalias genitais e um nariz rudimentar. O peito estreito e a morte precoce antes do décimo minuto de vida sugerem hipoplasia pulmonar grave. Este caso destaca a heterogeneidade fenotípica com um coloboma da pálpebra, hipertelorismo, exoftalmos e apêndices raros.
consideramos que o segundo caso no nosso estudo é uma síndrome tetramelia – 2 considerando a simetria tetra-amelia com a presença de cotos dos Membros Superiores. O diagnóstico de tetra-amelia deve ser feito precocemente durante o monitoramento de ultrassom. Por conseguinte, deve ser sensibilizada para a importância da monitorização por ultrassom e da utilização de 3D/4D para melhorar os resultados do rastreio. O diagnóstico de uma massa pélvis em ultrassom associada com a amelia deve levantar suspeitas de síndrome de defeito do membro de fusão espleno-gonadal.
além disso, o exame fetal e o teste fetal utilizando as tecnologias em evolução do microarray cromossómico e da sequenciação do Exoma e do genoma devem ser encorajados nos nossos cenários. Uma melhor caracterização dos casos permite dar conselhos aos casais e um melhor conhecimento dessas anomalias clínicas.
conclusão
a síndrome Tetra-amelia é escassa e as áreas cinzentas ainda permanecem. Estes dois casos, comparados com os já descritos na literatura, ilustram a heterogeneidade fenotípica da tetraamelia. Dada a rara incidência destas anomalias, seria importante criar um registo internacional de anomalias A fim de comunicar os casos e criar um banco de amostras para estudos genéticos alargados aos pais.
- Wilcox WR, Coulter CP, Schmitz ML (2015) Congenital limb deficiency disorders. Clin Perinatol 42: 281-300.
- Bermejo SE, Cuevas L, Amar e (2011) Amelia: a multi-center descriptive epidemiologic study in a large dataset from the International Clearinghouse for Birth Defects Surveillance and Research, and overview of the literature. Am J Med Genet C Semin Med Genet 157: 288-304.Zlotogora JSM, Shabany YO, Jarallah RY (1993) síndrome de tetraamelia com hipoplasia pulmonar. Am J Med Genet 47: 570-571.Zimmer EZ (1985) Tetra-amelia com malformações múltiplas em seis fetos masculinos de uma mesma família. Europ. J Pediat 144: 412-414.
- Rosenak D (1991) tetraamelia recorrente e hipoplasia pulmonar com malformações múltiplas em sibs. Am J Med Genet 38: 25-28.
- gershoni BR (1990) Roberts syndrome or “X-linked amelia” ? . Am J Med Genet 37: 569-572.Kosaki K, Jones MC, Stayboldt C (1996) Zimmer phocomelia: delineation by principal coordinate analysis. Am J Med Genet 66: 55-59.
- Niemann S (2004) a mutação homozigótica WNT3 causa tetra-amelia numa grande família consanguínea. Am J Hum Genet 74: 558-563.
- Krahn m (2005) Tetra-amelia and lung aplasia syndrome: report of a new family and exclusion of candidate genes. Clin Genet 68: 558-560.
- Szenker-RE, Altunoglu U (2018) a inibição RSPO2 da RNF43 e ZNRF3 rege o desenvolvimento dos Membros independentemente da LGR4/5/6. Nature 557: 564-569.