Paralyses periódicos
uma classificação clinicamente útil dos paralyses periódicos primários, mostrada na Tabela 1, inclui formas hipocaliémicas, hipercalémicas e paramiotónicas.
Quadro 1. Principal Periódico Paralisia (modificado a partir de Jurkat-Rott e Lehmann-Horn ) (Tabela Abrir em uma nova janela)
|
Doença |
Gene |
Proteína |
Herança |
Mutação |
|
HyperPP |
SCN4A |
Nav1.4 |
Dominante |
Multa |
|
NormoPP |
Multa (ω-poro) |
|||
|
Paramyotoniacongenita |
Multa |
|||
|
HypoPP Tipo II |
Multa (ω-poro) |
|||
|
HypoPP, A Fim de |
CACNA1S |
Cav1.1 |
Dominante |
Ganho (ω-poro) |
|
ThyrotoxicPP |
KCNJ18 |
Kir2.18 |
Dominante |
a Perda de |
|
Andersen-Tawil síndrome de |
KCNJ2 |
Kir2.1 |
Dominante |
a Perda de |
A base fisiológica da flácida fraqueza é inexcitability do músculo de membrana (ou seja, do sarcolema). A alteração do nível de potássio sérico não é o defeito principal na PP primária; o metabolismo de potássio alterado é um resultado da PP. No PP primário e tirotóxico, a paralisia flácida ocorre com alterações relativamente pequenas no nível de potássio sérico, enquanto no PP secundário, os níveis de potássio sérico são marcadamente anormais.
nenhum mecanismo único é responsável por este grupo de doenças. Assim, eles são heterogêneos, mas compartilham alguns traços comuns. A fraqueza geralmente é generalizada, mas pode ser localizada. A musculatura craniana e os músculos respiratórios são geralmente poupados. Os reflexos esticados ou estão ausentes ou diminuídos durante os ataques. As fibras musculares são eletricamente imperdoáveis durante os ataques. A força muscular é normal entre ataques, mas, após alguns anos, algum grau de fraqueza fixa se desenvolve em certos tipos de PP (especialmente PP primário). Todas as formas primárias de PP (exceto Becker myotonia congenita ) são herdadas autossômicas dominantes ou esporádicas (provavelmente decorrentes de mutações pontuais).
os canais iónicos sensíveis à tensão regulam de perto a geração de potenciais de Acção (alterações breves e reversíveis da tensão das membranas celulares). Estes são canais iônicos seletivamente e variavelmente permeáveis. Os transportadores de iões dependentes da energia mantêm gradientes de concentração. Durante a geração de potenciais de ação, iões de sódio se movem através da membrana através de canais iônicos de voltagem. A membrana da fibra muscular em repouso é polarizada principalmente pelo movimento do cloreto através dos canais de cloreto e é repolarizada pelo movimento do potássio. Sódio, cloreto e Canalizações de cálcio, como um grupo, estão associados com miotonia E PP. As subunidades funcionais dos canais de sódio, cálcio e potássio são homólogas. As canalizações de sódio são melhor compreendidas do que as canalizações de cálcio ou cloreto. Todas as formas de PP familiar mostram a via mecanicista final envolvendo despolarização aberrante, inactivação dos canais de sódio, e inexcitabilidade da fibra muscular.
a discussão neste artigo aborda principalmente as canalizações de sódio, cálcio e potássio, bem como formas secundárias de PP. Canalizações de cloreto não estão associadas com fraqueza episódica e são discutidos em mais detalhes nos artigos sobre distúrbios miotônicos.
Resumo de canal de disfunção em vários tipos de PP
Com HyperPP rápido canal de inativação, as mutações são normalmente situada na parte interna dos segmentos transmembrana ou no intracelular ciclos que afetam os locais de encaixe rápido inactivação de partículas, afetando rápido canal de inativação levando a persistente Na+ atual.
com uma fuga de catião activada por hipopp que contrai a corrente Rectificadora de K+, as mutações causam substituição da arginina ou da lisina.
com fuga de catião activada por despolarização NormoPP, as mutações estão em locais mais profundos do sensor de tensão do domínio II no codon R675.
a disfunção do canal iónico é geralmente bem compensada com excitação normal, e são frequentemente necessários gatilhos adicionais para produzir a inexcitabilidade muscular devido à despolarização contínua da membrana.A ingestão de Glucose e potássio tem os efeitos opostos nestas doenças. Na Hiperpp, a ingestão de potássio desencadeia o ataque, enquanto que a glucose a melhora. Em contraste, a glicose provoca ataques hipocalêmicos e potássio é o tratamento para o ataque.
Note A imagem abaixo.
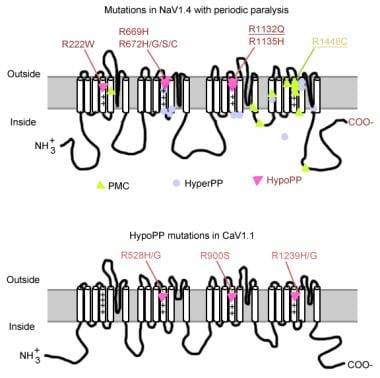 mutações na paralisia periódica.
mutações na paralisia periódica. gene do canal de sódio muscular
o canal de sódio tem uma subunidade alfa e uma subunidade beta. A subunidade alfa do canal de sódio é uma glicoproteína 260-kd que compreende cerca de 1800-2000 aminoácidos. Este canal é altamente conservado evolucionariamente de Drosophila para humano. Ele tem 4 homólogo domínios (I-IV), que dobra para formar um central de poro, cada um com 225-325 aminoácidos. Cada domínio é constituído de 6 segmentos hidrofóbicos (S1-S6) atravessar a membrana celular. As principais funções do canal incluem a catagem sensível à tensão, a inactivação e a selectividade iónica. O extracelular loop entre S5 e S6 mergulha na membrana plasmática e participa na formação do poro. O S4 segmento contém aminoácidos com carga positiva em todos terceira posição e funciona como um sensor de tensão. Mudanças de conformação podem ocorrer durante a despolarização, resultando na ativação e inativação do canal. O laço celular entre o domínio III-S6 e o domínio IV-S1 atua como uma porta inactiva.
o canal de sódio tem 2 portas (activação e inactivação) e pode existir em 3 estados. Em repouso com a membrana polarizada, o portal de ativação é fechado e o portal de inativação é aberto. Com a despolarização, o portal de ativação se abre, permitindo que íons de sódio passem através do canal de íons e também expondo um local de acoplagem para o portal de inativação. Com a despolarização contínua, a porta de inactivação fecha, bloqueando a entrada de sódio na célula e fazendo com que o canal entre no estado de inactivação rápida. Esta inativação do canal permite que a membrana se repolarize, resultando em um retorno ao estado de repouso com o portal de ativação fechado e o portão de inativação aberto. Dois processos de inactivação ocorrem no músculo esquelético dos mamíferos: a inactivação rápida envolve a interrupção do potencial de acção e actua numa escala de milissegundos de tempo. A inactivação lenta demora segundos a minutos e pode regular a população de canais de sódio excitáveis.
mutações nos canais de sódio que interrompem a inactivação rápida e lenta são geralmente associadas a um fenótipo de Hiperpp e miotonia, onde como mutações que aumentam a inactivação lenta ou rápida, produzindo perda da função dos canais de sódio, causam Hipopp.
as mutações do gene do canal de sódio (SCN4A) têm várias características gerais. A maioria das mutações estão no linker “inactivante” entre repetições III e IV, no segmento “sensor de voltagem” S4 da repetição IV ou na membrana interna, onde podem danificar o local de acoplagem para a porta de inactivação. O fenótipo clínico difere por substituição específica de aminoácidos e, embora alguma sobreposição possa ocorrer entre hipercalemia PP, paramiotonia congenita (PC), e miotonias agravadas pelo potássio (PAM), os 3 fenótipos são geralmente distintos (como descrito abaixo). Quase todos os canais mutantes prejudicaram a rápida inactivação da Corrente de sódio. A maioria dos pacientes são sensíveis ao potássio sistêmico ou à temperatura fria.
existem duas populações de canais, mutantes e de tipo selvagem; a inactivação rápida deficiente resulta numa despolarização prolongada das membranas mutantes da fibra muscular e pode explicar os 2 sintomas cardinais destas doenças, miotonia e fraqueza. Em hipercalemia PP, um ganho de função ocorre na mistura de canais mutantes, resultando em um aumento da Corrente de sódio excessivamente despolarizando o músculo afetado. A despolarização ligeira (5-10 mV) da membrana da miofiber, que pode ser causada pelo aumento das concentrações de potássio extracelular, resulta na manutenção dos canais mutantes no modo não inactivado. A corrente de sódio interna persistente causa o disparo repetitivo dos canais de sódio de tipo selvagem, que é percebido como rigidez (ou seja, miotonia).
se estiver presente uma despolarização mais grave (20-30 mV), tanto os canais normais como os anormais são fixados num estado de inactivação, causando fraqueza ou paralisia. Assim, diferenças sutis na gravidade da despolarização da membrana podem fazer a diferença entre miotonia e paralisia. A sensibilidade à temperatura é uma característica do PC. O frio exacerba a miotonia e induz fraqueza. Um número de mutações estão associadas a esta condição, 3 delas no mesmo local (1448) no segmento S4. Estas mutações substituem a arginina por outros aminoácidos e neutralizam esta carga positiva S4 altamente conservada. Mutações destes resíduos são a causa mais comum de PC. Alguns dos possíveis mecanismos responsáveis pela sensibilidade à temperatura incluem o seguinte::
-
a temperatura Pode afectar diferentemente a alteração conformacional do canal mutante.
-
temperaturas mais baixas podem estabilizar os canais mutantes num estado anormal.
-
mutações podem alterar a sensibilidade do canal a outros processos celulares, tais como fosforilação ou segundo mensageiro.
a maioria dos casos de hipercaliemia PP é devida a 2 mutações em SCN4A, T704M e M1592V. Mutações no canal de sódio, especialmente nos resíduos 1448 e 1313, são responsáveis pela paramiotonia congenita. Uma pequena proporção de casos de paralisia periódica hipocaliêmica está associada com mutações nos codões 669 e 672 (HypoPP2). No Hipopp2, as mutações dos canais de sódio aumentam a inactivação para produzir uma perda líquida de defeito da função.
A PP Normocalémica assemelha-se tanto à Hiperpp (sensibilidade ao potássio) como à Hipopp (duração dos ataques) e é causada por mutações SCN4A numa localização mais profunda do sensor de tensão DII no codon 675. R675 mutações diferem de HypoPP na medida em que estas mutações resultam em Poros geradores de ω-corrente activados pela despolarização, com dependência de voltagem invertida, uma vez que este local é exposto a locais extracelulares com uma despolarização mais forte.
gene do canal de cálcio
o gene do canal de cálcio (CACNL1A3) é um complexo de 5 subunidades (alfa-1, alfa-2, beta, gama e delta). O receptor da dihidropiridina (DHP) do músculo esquelético está localizado principalmente na membrana tubular transversal. A subunidade alfa-1 tem locais de ligação para drogas DHP e conduz a corrente L-tipo de cálcio lenta. Ele também participa do acoplamento excitação-contração (CE) e atua como um sensor de tensão através de sua ligação com o receptor de ryanodina reticulum sarcoplasmático (ou seja, canal de liberação de cálcio). Quaisquer alterações no potencial da membrana estão ligadas à libertação intracelular de cálcio, permitindo o acoplamento CE. Mutações pontuais na subunidade alfa-1 do receptor de DHP/canal de cálcio causam hipocaliemia PP (Hipopp1). Duas mutações do gene CACNA1S, R528H e R1239H, são responsáveis pela maioria dos casos de PP hipocaliémico.
a base fisiológica da doença ainda não é compreendida, mas é mais provável devido a uma falha de excitação em vez de uma falha do acoplamento CE. No entanto, a despolarização induzida pela hipocaliemia pode reduzir a libertação de cálcio, afectando o controlo de tensão do canal, directa ou indirectamente, através da inactivação do canal de sódio. A insulina e a adrenalina podem actuar de forma semelhante. As mutações do gene do canal de cálcio têm algumas semelhanças com as mutações SCN4A. As mutações modificam a inactivação do canal, mas não a activação dependente da voltagem. Gravações de culturas de myotube de pacientes afetados revelaram uma redução de 30% na corrente L-tipo de cálcio sensível ao DHP. Os canais são inactivados a potenciais de membrana Baixos.
mutações nos canais de cálcio provocam uma perda de função manifestada como uma densidade de corrente reduzida e uma inactivação mais lenta. Não se compreende como esta inactivação está relacionada com ataques induzidos por hipocaliemia. Pelo menos na mutação R528H, ocorre uma possível channelopatia secundária, ligada a uma redução da Corrente de potássio sensível ao ATP a partir da homeostase de cálcio alterada. As correntes mais baixas associadas às mutações CACNL1A3 podem alterar ligeiramente a homeostase intracelular do cálcio, o que pode afectar as propriedades e expressão dos canais K+, particularmente KATP (canal de potássio sensível ao ATP) pertencente à classe retifier interna dos canais. A insulina actua também em Hipopp, reduzindo este retificador K+ actual.
a perda de carga do sensor de tensão é responsável pela maioria dos casos de Hipopp. Os canais de sódio e cálcio têm subunidades alfa formadoras de poros homólogas. As mutações pontuais em CACNL1A3 e SCN4A afetam os resíduos argentinos nos sensores de tensão S4 destes canais. As mutações de arginina nos segmentos S4 são responsáveis por 90% dos casos de Hipopp.
a perda de carga do sensor de tensão é responsável pela maioria dos casos de Hipopp. Os canais de sódio e cálcio têm subunidades α formadoras de poros homólogas. Quase todas as mutações em Cav1.1 (HypoPP-1) e Nav1.4 (HypoPP-2) neutralizam um aminoácido carregado positivamente em uma das argininas ou lisinas mais exteriores dos sensores de tensão. O Nav1.4 mutações são mais comumente situadas nos sensores de tensão de I, II e III repetições, causando uma fuga de catião.
Substituição de ultraperiféricas arginina com uma menor de aminoácidos como glicina abre uma condutora do caminho ao hyperpolarized potencial, resultando em um interior cação atual (cação de fuga ou ω corrente de distinguir (ω-) através de íon–realização de poros, é uma hiperpolarização-ativado atual de catiões monovalentes através S4 disparo de poros contrariando a rectificação K+ correntes) despolarização ou desestabilizar o potencial de repouso.
S4 segment moves outward during depolarization closing the conductive pathway. Fibras musculares com graves mutações de sensores de tensão são despolarizadas não só durante a hipocaliemia, mas também em níveis de potássio no intervalo normal, explicando a fraqueza interictal e permanente. A miopatia grave com substituição de gordura do tecido muscular é frequentemente encontrada em doentes com mutações Cav1.1 R1239H (mutações DIV). Os glucocorticosteróides causam Hipopp estimulando a Na+ K+ ATPase mediada pela insulina e amilina.
gene do canal de potássio
retificação interna é uma propriedade importante dos canais Kir. A retificação envolve o bloqueio de poros dependentes da tensão com poliaminas e Mg++ durante a despolarização, e este bloqueio é removido durante o gradiente potencial durante a hiperpolarização. Observam-se mutações nos canais de potássio na síndrome de Andersen-Tawil e na PP tirotóxica.
a tríade de características dismórficas, paralisia periódica e arritmias cardíacas caracterizam a síndrome de Andersen-Tawil. Esta síndrome está associada a mutações no gene KCNJ2. O gene KCNJ2 codifica o canal de potássio Kir2.1. As mutações dos canais de potássio no KCNE3 causam hipocaliemia PP, mas isto não foi fundamentado.
mutações em Kir2. 6 causam susceptibilidade à PP tirotóxica. A fraqueza episódica observada na PP tirotóxica é semelhante à observada na Hipopp e na síndrome de Andersen-Tawil. Esta desordem é mais prevalente nos asiáticos e nos homens latino-americanos. A tirotóxica PP é uma doença genética desmascarada pela tirotoxicose. Kir2. 6 é principalmente expresso em músculo esquelético. A triiodotironina aumenta a transcrição KCNJ18, que pode conduzir a expressão melhorada de Kir2. 6. A PKC é activada durante a tirotoxicose devido ao aumento da rotatividade do PIP2 e os canais Kir interagem directamente com o PIP2 durante a concentração normal. Na síndrome de Andersen-Tawil, há diminuição da afinidade do PIP2. No PP tirotóxico, nenhuma das mutações altera a retificação de Kir2.6.