ginecologie și obstetrică raport de caz
cuvinte cheie
Tetra-amelia; malformație; genotip; fenotip.
Introducere
anomaliile membrelor constituie un grup important de patologii congenitale caracterizate prin hipoplazie sau aplazie completă a unuia sau mai multor oase ale membrelor. Anomaliile membrelor de toate tipurile apar la aproximativ 1 din 1.300 până la 2.000 de nașteri. Aceste anomalii ale membrelor pot fi izolate sau asociate cu alte anomalii . Sindromul Tetraamelia este rar și rămân zone gri.
raportăm două cazuri de tetra-amelia într-o maternitate de nivel II din Dakar (Senegal) cu a fi similar cu tetraamelia-1 (cromozomul 17q21), tetraamelia-2 (cromozomul 8q23) și sindromul Robert (cromozomul 8p21). Acest lucru ilustrează dificultatea corelării fenotipului și a genelor implicate.
rapoarte de caz
cazul 1
Doamna AD a fost o mamă de 44 de ani trimisă la departamentul nostru la 36 de săptămâni de gestație cu preeclampsie severă și anomalii fetale. Ea a fost de cinci para cu nici un istoric de anomalii fetale. Nu fuma acum și niciodată nu fumase și nici nu băuse alcool. Nu a fost expusă fumatului pasiv. Era într-o căsătorie consanguină de gradul trei pentru toți copiii ei. Dna AD a avut rezultate negative pentru hepatita B, HIV și sifilis. Ea a fost protejată de virusul rubeolei și nu a avut expunere anterioară la Toxoplasma gondii. Monitorizarea cu ultrasunete efectuată cu întârziere la 33 săptămâni și 35 săptămâni de gestație a recuperat oligoamnioză și hidrocefalie, precum și ageneză a membrelor. Prescripțiile în timpul sarcinii au inclus administrarea de fier și acid folic, precum și administrarea de pirimetamină sulfadoxină. Acesta din urmă a fost prescris la 18 săptămâni și apoi la 26 de săptămâni ca parte a politicii de profilaxie anti-malarie pentru femeile însărcinate. Înălțimea simfizală-fundală a măsurat 28 cm. Datorită caracteristicilor severe ale preeclampsiei; a fost spitalizată imediat și observată într-o unitate de muncă și livrare. Apoi a primit inițial sulfat de magneziu IV pentru a preveni eclampsia și medicamente antihipertensive pentru a menține tensiunea arterială sistolică sub 160 mmHg și tensiunea arterială diastolică sub 105 mmHg.
a fost luată decizia de livrare imediată prin cezariană. A fost extras un 2.150 de grame născut viu, care ulterior a murit în decurs de 10 minute. Înainte de moarte, corpul a prezentat mișcări târâtoare. Au fost identificate mai multe anomalii externe (Figura 1) incluzând ageneza completă a tuturor celor patru membre, hidrocefalie cu circumferința capului de 39 cm. Pe față, a existat hipertelorism cu un colobom al pleoapelor, exoftalmie ușoară și aniridia. Gura era la fel un V inversat fără o delimitare clară a buzelor, iar nasul era rudimentar. Nou-născutul era lipsit de tegumente (păr și sprâncene). Urechile au fost reduse la schițe care arătau ca niște fante. Gâtul era scurt. Trunchiul a fost redus la o structură conică lungă de 26 cm, cu un cordon ombilical la capătul inferior. Pieptul era îngust. Chiar sub buric, trunchiul lărgit înapoi era prezent la podeaua polului caudal, o linie mediană cu recesiuni și o înmugurire care poate corespunde unui falus de tip nedeterminat. Au fost observate ageneza pelvisului, a organelor genitale și a imperforării anale. Patologia fetală nu a fost efectuată. Cu toate acestea, moartea în decurs de 10 minute de la naștere și aspectul conic al pieptului pot sugera anomalii pulmonare.
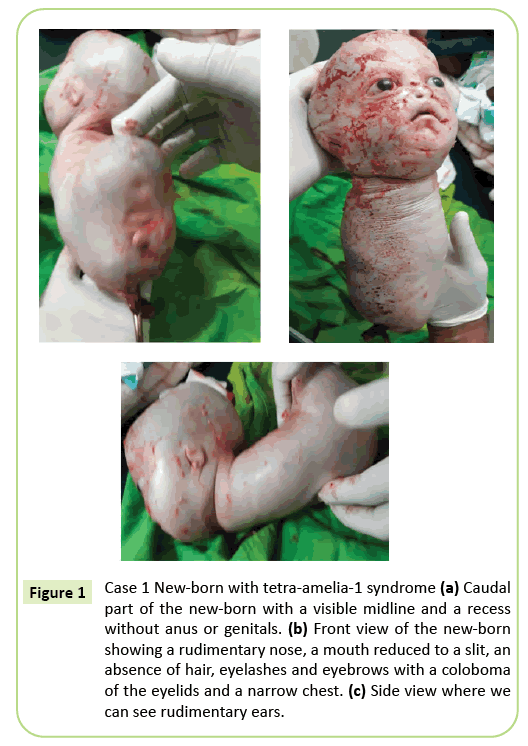
Figura 1: Cazul 1 nou-născut cu sindromul tetra-amelia-1 (a) parte caudală a nou-născutului cu o linie mediană vizibilă și o adâncitură fără anus sau organe genitale. (b) vedere frontală a nou-născutului care prezintă un nas rudimentar, o gură redusă la o fantă, o absență a părului, genelor și sprâncenelor cu un colobom al pleoapelor și un piept îngust. (c) vedere laterală unde putem vedea urechi rudimentare.
Cazul 2
al doilea caz a fost un primigravida în vârstă de 22 de ani, trimis la unitatea noastră pentru scanare cu ultrasunete la 37 de săptămâni de gestație. Nu era într-o căsătorie consangvină. Ea a testat negativ pentru hepatita B, HIV și sifilis. Nu a fost testată pentru toxoplasmoză și rubeolă. Nu s-a efectuat nicio monitorizare cu ultrasunete în timpul sarcinii. Examenul clinic a fost în concordanță cu întârzierea creșterii fetale (înălțimea fundului: 26 cm). Rezultatele cu ultrasunete au arătat că humerusul este distorsionat măsurând 23,9 mm corespunzând la 17 săptămâni de gestație. A fost agenezie a femurului. Aripile iliace erau vizibile pe ultrasunete. Nu au fost identificate anomalii pulmonare sau cardiace. Livrarea a fost inițiată. Nou-născutul a avut un fenotip feminin cu un scor Apgar de 9 în minutul 5. Morfologia capului și a trunchiului a fost fără particularitate. Membrele superioare au fost reduse la două cioturi lungi de 3 cm. A fost observată ageneza completă a celor 2 membre inferioare. A fost o anomalie simetrică (Figura 2).
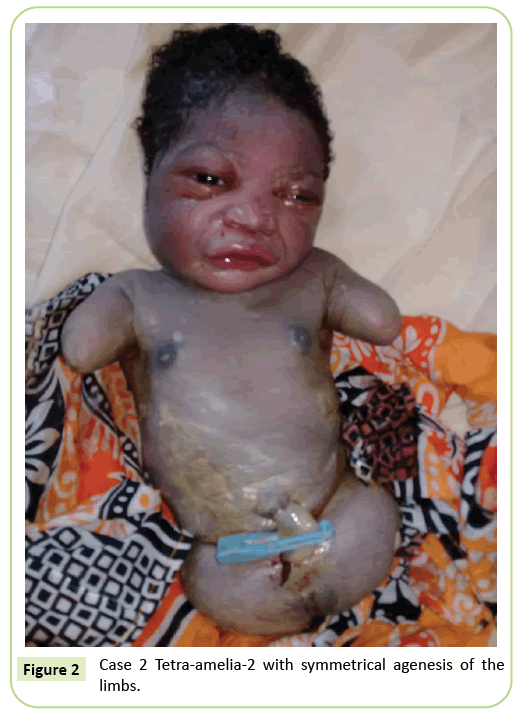
Figura 2: Cazul 2 Tetra-amelia-2 cu ageneză simetrică a membrelor.
discuție
Bermejo-Sanchez și colab. a descris în 2011 epidemiologia Ameliei congenitale folosind date colectate din 20 de programe de supraveghere a anomaliilor congenitale, de pe toate continentele, cu excepția Africii, între 1968 și 2006. În total, au fost identificate 326 de cazuri de amelia dintre 23.110.591 de nașteri vii, nașteri morți și avorturi. Prevalența a fost de 1,41/100.000 .
Tetra-amelia se referă la absența completă a membrelor și apare mai rar. Din cunoștințele noastre, tetra-amelia-1 este descrisă în 7 familii. Se pare că urmează o moștenire autosomală recesivă. În toate familiile, tetra-amelia-1 a fost asociată cu malformații severe ale celorlalte părți ale corpului, inclusiv fața și capul, anomalii ale sistemului nervos, scheletului și organelor genitale. Plămânii au fost subdezvoltați în multe cazuri, ceea ce face respirația dificilă sau imposibilă . Zimmer și colab. raportat în 1985 o familie puternic consangvinizată în care 6 sugari au avut tetra-amelia-1 și hidrocefalie. Ei au descris într-unul dintre fetuși o absență totală a osului pelvian, a buzei și a palatului, a arrhiniei și a aplaziei urechilor. S-a observat, de asemenea, un plămân stâng bilateral, un canal arterial persistent, o imperforare anală. Testarea fetală a eliminat diagnosticul sindromului Robert . Alte cazuri recuperate în literatură includ cel al lui Kosaki și colab., în 1996, cu un făt de cariotip 46, XX cu tetrafocomelie și hipoplazie pulmonară severă, pe lângă anomaliile feței și capului . Rosenak și colab. a descris un caz de tetra-Amelie cu hipoplazie pulmonară severă la doi fetuși ai unui cuplu non-consanguin. Testele fetale au exclus diagnosticul sindromului Robert . Două cazuri suplimentare au fost raportate de Zlotogora și colab. în 1993. Ambii pacienți au murit la scurt timp după naștere, iar autorii au sugerat existența hipoplaziei pulmonare. Niemann și colab. a raportat o familie turcă consanguină în care 4 din cei 8 frați sufereau de tetra-amelia. În plus față de absența celor 4 membre, examinările fetale ale 3 fetuși au evidențiat multiple anomalii: buzele despicate și /sau palatine, laparoschisis, anomalii pulmonare, hipoplazia pelvisului, atrezia choanelor, vaginul și imperforarea anală . În cele din urmă, în 2005, Krahn și colab. descris 2 frați născuți din părinți consangvinizați care suferă de tetraamelie și hipoplazie pulmonară severă. Claviculele și omoplații au fost normale la al doilea făt. Cariotipul era normal .
sindromul Tetra-amelia-1 sau TETAMS1 este cauzat de o mutație homozigotă în gena WNT3 pe cromozomul 17q21 cu o moștenire autosomală recesivă. Sindromul Tetraamelia – 2 (TETAMS2) se caracterizează prin membre rudimentare sau absența completă a membrelor, în general ageneză simetrică și bilaterală a plămânilor în unele cazuri. Sunt, de asemenea, anomalii obișnuite ale sistemului vascular pulmonar și dismorfii incluzând buza și Palatul bilateral, anchiloglossia, hipoplazia mandibulară, microretrognatia și aplazia labioscrotală .
Szenker-Ravi, studiind 4 familii de tetra-amelia cu ageneză sau hipoplazie pulmonară, a observat o eterogenitate fenotipică cu anomalii ale membrelor de severitate variabilă . Secvențierea exomului în aceste 4 familii a făcut posibilă identificarea mutațiilor homozigote trunchiate în gena RSPO2 . Sindromul Tetraamelia – 2 este cauzat de o mutație homozigotă în gena RSPO2 (610575) localizată pe cromozomul 8q23 .
fenotipul primului caz descris în acest articol corespunde unui sindrom tetra-amelia-1 datorat în special prezenței hidrocefaliei, anomaliilor genitalia și un nas rudimentar. Pieptul îngust și moartea timpurie înainte de minutul 10 al vieții sugerează hipoplazie pulmonară severă. Acest caz evidențiază eterogenitatea fenotipică cu un colobom al pleoapelor, hipertelorism, exoftalmie și anexe rare.
considerăm că al doilea caz în studiul nostru este un sindrom tetramelia-2 având în vedere tetra-amelia simetrică cu prezența buturugilor membrelor superioare. Diagnosticul tetra-Ameliei trebuie făcut devreme în timpul monitorizării cu ultrasunete. Prin urmare, ar trebui să se conștientizeze importanța monitorizării cu ultrasunete și utilizarea 3D/4D pentru a îmbunătăți rezultatele screeningului. Diagnosticul unei mase pelviene pe ultrasunete asociat cu amelia ar trebui să ridice suspiciunea pentru sindromul defectului membrelor de fuziune spleno-gonadală.
în plus, examinarea fetală și testarea fetală folosind tehnologiile în evoluție ale microarray-ului cromozomial și secvențierea exomului și a genomului trebuie încurajate în setările noastre. O mai bună caracterizare a cazurilor face posibilă oferirea de sfaturi cuplurilor și o mai bună cunoaștere a acestor anomalii clinice.
concluzie
sindromul Tetra-amelia este rar și zonele gri rămân în continuare. Aceste două cazuri, în comparație cu ceea ce este deja descris în literatură, ilustrează eterogenitatea fenotipică a tetraameliei. Având în vedere incidența rară a acestor anomalii, ar fi important să se creeze un registru internațional al anomaliilor pentru a raporta cazurile și a înființa o bancă de eșantioane pentru studii genetice extinse la părinți.
- Wilcox WR, Coulter CP, Schmitz ML (2015) tulburări congenitale ale deficienței membrelor. Clin Perinatol 42: 281-300.
- Bermejo se, Cuevas L, Amar e (2011) Amelia: un studiu epidemiologic descriptiv multi-centru într-un set mare de date de la International Clearinghouse pentru Supravegherea și cercetarea defectelor congenitale și o prezentare generală a literaturii. Am J Med Genet C Semin Med Genet 157: 288-304.
- Zlotogora JSM, Shabany YO, Jarallah RY (1993) sindromul tetraameliei cu hipoplazie pulmonară. Am J Med Genet 47: 570-571.
- Zimmer EZ (1985) Tetra-amelia cu malformații multiple la șase fetuși masculi într-o singură rudă. Europ. J Pediat 144: 412-414.
- Rosenak D (1991) tetraamelia recurentă și hipoplazia pulmonară cu malformații multiple la sibs. Am J Med Genet 38: 25-28.
- Gershoni BR (1990) sindromul Roberts sau „amelia legată de X” ? . Am J Med Genet 37: 569-572.
- Kosaki K, Jones MC, Stayboldt C (1996) Zimmer phocomelia: delimitarea prin analiza coordonatelor principale. Am J Med Genet 66: 55-59.
- Niemann S(2004) mutația homozigotă wnt3 provoacă tetra-amelia într-o mare familie consanguină. Am J Hum Genet 74: 558-563.
- Krahn M (2005)sindromul Tetra-amelia și aplazia pulmonară: raportul unei noi familii și excluderea genelor candidate. Clin Genet 68: 558-560.
- Szenker-RE, Altunoglu U (2018) inhibarea RSPO2 a RNF43 și ZNRF3 guvernează dezvoltarea membrelor independent de LGR4/5/6. Natură 557: 564-569.