paralizii periodice
o clasificare utilă clinic a paraliziilor periodice primare, prezentată în tabelul 1, include formele hipokalemice, hiperkalemice și paramiotonice.
Tabelul 1. Paralizie periodică primară (modificată de la Jurkat-Rott și Lehmann-Horn ) (Masă deschisă într-o fereastră nouă)
|
boala |
Gene |
proteine |
moștenire |
mutație |
|
HyperPP |
SCN4A |
Nav1.4 |
dominante |
bine |
|
NormoPP |
Fine (cu pori-de-la-La-La-La) |
|||
|
Paramyotoniacongenita |
bine |
|||
|
Hipopp tip II |
Fine (cu pori-de-la-La-La-La) |
|||
|
Hipopp pentru a |
CACNA1S |
Cav1.1 |
dominante |
Gain (cu pori) |
|
Tirotoxicp |
KCNJ18 |
Kir2.18 |
dominante |
pierdere |
|
sindromul Andersen-Tawil |
KCNJ2 |
Kir2.1 |
dominante |
pierdere |
baza fiziologică a slăbiciunii flasc este inexcitabilitatea membranei musculare (adică sarcolema). Modificarea concentrației plasmatice a potasiului nu este principalul defect al PP primar; metabolizarea modificată a potasiului este rezultatul PP. În PP primar și tirotoxic, paralizia flască apare cu modificări relativ mici ale nivelului seric de potasiu, în timp ce în PP secundar, nivelurile serice de potasiu sunt semnificativ anormale.
niciun mecanism unic nu este responsabil pentru acest grup de tulburări. Astfel, ele sunt eterogene, dar împărtășesc unele trăsături comune. Slăbiciunea este de obicei generalizată, dar poate fi localizată. Mușchii cranieni și mușchii respiratori sunt de obicei scutiți. Reflexele de întindere sunt fie absente, fie diminuate în timpul atacurilor. Fibrele musculare sunt inexcitabile electric în timpul atacurilor. Forța musculară este normală între atacuri, dar, după câțiva ani, se dezvoltă un anumit grad de slăbiciune fixă în anumite tipuri de PP (în special PP primar). Toate formele de PP primar (cu excepția Becker myotonia congenita) sunt fie autosomal dominant moștenit sau sporadic (cel mai probabil care rezultă din mutații punctuale).
canalele ionice sensibile la tensiune reglează îndeaproape generarea potențialelor de acțiune (modificări scurte și reversibile ale tensiunii membranelor celulare). Acestea sunt canale ionice permeabile selectiv și variabil. Transportorii de ioni dependenți de energie mențin gradienții de concentrație. În timpul generării potențialelor de acțiune, ionii de sodiu se deplasează prin membrană prin canale ionice cu tensiune. Membrana fibrelor musculare de repaus este polarizată în primul rând prin mișcarea clorurii prin canalele de clorură și este repolarizată prin mișcarea potasiului. Canalele de sodiu, clorură și calciu, ca grup, sunt asociate cu miotonia și PP. Subunitățile funcționale ale canalelor de sodiu, calciu și potasiu sunt omoloage. Channelopatiile de sodiu sunt mai bine înțelese decât channelopatiile de calciu sau clorură. Toate formele de PP familial arată calea mecanică finală care implică depolarizarea aberantă, inactivarea canalelor de sodiu și inexcitabilitatea fibrelor musculare.
discuția din acest articol abordează în primul rând canalopatiile de sodiu, calciu și potasiu, precum și formele secundare ale PP. Channelopatiile de clorură nu sunt asociate cu slăbiciune episodică și sunt discutate mai detaliat în articolele despre tulburările miotonice.
Rezumatul disfuncției canalului în diferite tipuri de PP
cu inactivarea rapidă a canalului Hiperpp, mutațiile sunt de obicei situate în părțile interioare ale segmentelor transmembranare sau în buclele intracelulare care afectează locurile de andocare pentru particula de inactivare rapidă, afectând astfel inactivarea rapidă a canalului, ducând la persistența curentului Na+.
cu hipopp hiperpolarizare-activat cation scurgere contracararea k + – curent de rectificare, mutatii provoca arginina ultraperiferice sau substituție lizină.
cu scurgerea cationică activată de depolarizare NormoPP, mutațiile se află în locații mai profunde ale senzorului de tensiune al domeniului II la codonul R675.
disfuncția canalului ionic este de obicei bine compensată cu excitație normală, iar declanșatorii suplimentari sunt adesea necesari pentru a produce inexcitabilitate musculară datorită depolarizării susținute a membranei.
aportul de glucoză și potasiu are efecte opuse în aceste tulburări. În Hiperpp, aportul de potasiu declanșează atacul, în timp ce glucoza îl ameliorează. În schimb, glucoza provoacă atacuri hipokalemice, iar potasiul este tratamentul atacului.
notați imaginea de mai jos.
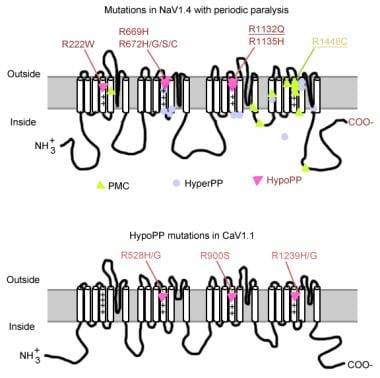 mutații în paralizia periodică.
mutații în paralizia periodică. Gena canalului de sodiu muscular
canalul de sodiu are o subunitate alfa și o subunitate beta. Subunitatea alfa a canalului de sodiu este o glicoproteină de 260 KD care cuprinde aproximativ 1800-2000 aminoacizi. Acest canal este foarte conservat evolutiv de la Drosophila la om. Are 4 domenii omoloage (I-IV) care se pliază pentru a forma un por central, fiecare cu 225-325 aminoacizi. Fiecare domeniu este format din 6 segmente hidrofobe (S1-S6) care traversează membrana celulară. Principalele funcții ale canalului includ închiderea sensibilă la tensiune, inactivarea și selectivitatea ionilor. Bucla extracelulară dintre S5 și S6 se scufundă în membrana plasmatică și participă la formarea porilor. Segmentul S4 conține aminoacizi încărcați pozitiv la fiecare a treia poziție și funcționează ca un senzor de tensiune. Modificările conformației pot apărea în timpul depolarizării, ducând la activarea și inactivarea canalului. Bucla celulară dintre domeniul III-S6 și domeniul IV-S1 acționează ca o poartă de inactivare.
canalul de sodiu are 2 porți (activare și inactivare) și poate exista în 3 stări. În repaus cu membrana polarizată, poarta de activare este închisă și poarta de inactivare este deschisă. Odată cu depolarizarea, poarta de activare se deschide, permițând ionilor de sodiu să treacă prin canalul ionic și, de asemenea, expunând un loc de andocare pentru poarta de inactivare. Cu depolarizarea continuă, poarta de inactivare se închide, blocând intrarea sodiului în celulă și determinând intrarea canalului în starea de inactivare rapidă. Această inactivare a canalului permite membranei să se repolarizeze, rezultând o revenire la starea de repaus cu poarta de activare închisă și poarta de inactivare deschisă. Două procese de inactivare apar în mușchiul scheletic al mamiferelor: inactivarea rapidă implică încetarea potențialului de acțiune și acționează pe o scară de timp de milisecunde. Inactivarea lentă durează secunde până la minute și poate regla populația canalelor de sodiu excitabile.
mutațiile canalului de sodiu care perturbă inactivarea rapidă și lentă sunt de obicei asociate cu un fenotip de Hiperpp și miotonie, unde mutațiile care sporesc inactivarea lentă sau rapidă produc pierderea funcției canalului de sodiu provoacă Hipopp.
mutațiile genei canalului de sodiu (SCN4A) au mai multe caracteristici generale. Majoritatea mutațiilor se află în legătura” inactivantă „dintre repetițiile III și IV, în segmentul” senzor de tensiune ” S4 al repetării IV sau la membrana interioară unde ar putea afecta locul de andocare pentru poarta de inactivare. Fenotipul clinic diferă prin substituția specifică a aminoacizilor și, în timp ce pot apărea unele suprapuneri între pp hiperkalemic, paramyotonia congenita (PC) și miotonii agravate de potasiu (PAM), cele 3 fenotipuri sunt în general distincte (așa cum este descris mai jos). Aproape toate canalele mutante au afectat inactivarea rapidă a curentului de sodiu. Majoritatea pacienților sunt sensibili la potasiul sistemic sau la temperatura rece.
există două populații de canale, de tip mutant și sălbatic; inactivarea rapidă afectată are ca rezultat depolarizarea prelungită a membranelor fibrelor musculare mutante și poate explica cele 2 simptome cardinale ale acestor tulburări, miotonia și slăbiciunea. În PP hiperkalemic, un câștig de funcție apare în închiderea canalului mutant, rezultând un curent crescut de sodiu depolarizând excesiv mușchiul afectat. Depolarizarea ușoară (5-10 mV) a membranei miofibre, care poate fi cauzată de creșterea concentrațiilor extracelulare de potasiu, are ca rezultat menținerea canalelor mutante în modul neinactivat. Curentul persistent de sodiu interior determină arderea repetitivă a canalelor de sodiu de tip sălbatic, care este percepută ca rigiditate (adică miotonie).
dacă este prezentă o depolarizare mai severă (20-30 mV), atât canalele normale, cât și cele anormale sunt fixate într-o stare de inactivare, provocând slăbiciune sau paralizie. Astfel, diferențele subtile în severitatea depolarizării membranei pot face diferența între miotonie și paralizie. Sensibilitatea la temperatură este un semn distinctiv al PC-ului. Rece exacerbează miotonia și induce slăbiciune. O serie de mutații sunt asociate cu această afecțiune, 3 dintre ele în același loc (1448) în segmentul S4. Aceste mutații înlocuiesc arginina cu alți aminoacizi și neutralizează această sarcină pozitivă S4 foarte conservată. Mutațiile acestor reziduuri sunt cea mai frecventă cauză a PC. Unele dintre mecanismele posibile responsabile de sensibilitatea la temperatură includ următoarele:
-
temperatura poate afecta diferențiat schimbarea conformațională a canalului mutant.
-
temperaturile mai scăzute pot stabiliza canalele mutante într-o stare anormală.
-
mutațiile pot modifica sensibilitatea canalului la alte procese celulare, cum ar fi fosforilarea sau al doilea mesager.
majoritatea cazurilor de PP hiperkalemic se datorează a 2 mutații în SCN4A, T704M și m1592v. Mutațiile din canalul de sodiu, în special la reziduurile 1448 și 1313, sunt responsabile pentru paramyotonia congenita. O mică parte din cazurile de paralizie periodică hipokalemică sunt asociate cu mutații la codonii 669 și 672 (Hipopp2). În Hipopp2, mutațiile canalului de sodiu sporesc inactivarea pentru a produce o pierdere netă a defectului funcției.
Normokalemic PP seamănă atât cu Hiperpp (sensibilitate la potasiu), cât și cu Hipopp (durata atacurilor) și este cauzată de mutații SCN4A la o locație mai profundă a senzorului de tensiune DII la codonul 675. Mutațiile R675 diferă de Hipopp prin faptul că aceste mutații au ca rezultat activarea depolarizării porilor de închidere generând curent de curent cu tensiune inversată, deoarece acest situs este expus siturilor extracelulare la o depolarizare mai puternică.
Gena canalului de calciu
gena canalului de calciu (CACNL1A3) este un complex de 5 subunități (alfa-1, alfa-2, beta, gamma și delta). Receptorul dihidropiridinei musculare scheletice (DHP) este localizat în principal în membrana tubulară transversală. Subunitatea alfa-1 are situsuri de legare pentru medicamentele DHP și conduce curentul lent de calciu de tip L. De asemenea, participă la cuplarea excitație-contracție (ce) și acționează ca un senzor de tensiune prin legătura sa cu receptorul ryanodină al reticulului sarcoplasmic (adică canalul de eliberare a calciului). Orice modificare a potențialului membranei este legată de eliberarea intracelulară de calciu, permițând cuplarea ce. Mutațiile punctuale în subunitatea alfa-1 a receptorului DHP/canalului de calciu determină PP hipokalemic (HypoPP1). Două mutații ale genei CACNA1S, R528H și R1239H, sunt responsabile pentru majoritatea cazurilor de PP hipokalemic.
baza fiziologică a bolii nu este încă înțeleasă, dar este mai probabil din cauza unei defecțiuni a excitației, mai degrabă decât a unei defecțiuni a cuplării ce. Cu toate acestea, depolarizarea indusă de hipokaliemie poate reduce eliberarea de calciu, afectând controlul tensiunii canalului direct sau indirect prin inactivarea canalului de sodiu. Insulina și adrenalina pot acționa în mod similar. Mutațiile genei canalului de calciu au unele asemănări cu mutațiile SCN4A. Mutațiile modifică inactivarea canalului, dar nu activarea dependentă de tensiune. Înregistrările din culturile myotube de la pacienții afectați au evidențiat o reducere de 30% a curentului de calciu de tip L sensibil la DHP. Canalele sunt inactivate la potențiale scăzute ale membranei.
mutațiile canalelor de calciu determină o pierdere a funcției manifestată ca o densitate redusă a curentului și o inactivare mai lentă. Modul în care această inactivare este legată de atacurile induse de hipokaliemie nu este înțeleasă. Cel puțin în mutația R528H, apare o posibilă channelopatie secundară, legată de o reducere a curentului de potasiu sensibil la ATP din homeostazia calciului modificată. Curenții inferiori asociați cu mutațiile CACNL1A3 ar putea modifica ușor homeostazia intracelulară a calciului, ceea ce ar putea afecta proprietățile și expresia canalelor K+, în special KATP (canalul de potasiu sensibil la ATP) aparținând clasei de canale redresoare interioare. Insulina acționează, de asemenea, în Hipopp prin reducerea acestui redresor interior k+ curent.
pierderile de încărcare ale senzorului de tensiune reprezintă majoritatea cazurilor de Hipopp. Canalele de sodiu și calciu au subunități alfa omoloage care formează pori. Mutațiile punctuale din CACNL1A3 și SCN4A afectează reziduurile argentiniene în senzorii de tensiune S4 ai acestor canale. Mutațiile argininei în segmentele S4 sunt responsabile pentru 90% din cazurile de Hipopp.
pierderile de încărcare ale senzorului de tensiune reprezintă majoritatea cazurilor de Hipopp. Canalele de sodiu și calciu au subunități omoloage de formare a porilor. Aproape toate mutațiile din Cav1.1 (Hipopp-1) și Nav1.4 (Hipopp-2) neutralizează un aminoacid încărcat pozitiv într-una dintre argininele sau lizinele exterioare ale senzorilor de tensiune. Nav1.4 mutații sunt cel mai frecvent situate în senzorii de tensiune ai repetițiilor I, II și III, provocând o scurgere de cationi.
Substituirea argininei ultraperiferice cu un aminoacid mai mic, cum ar fi glicina, deschide o cale conductivă la potențialul hiperpolarizat, rezultând un curent cationic interior (scurgere de cationi sau curent de la un curent de la un curent de la un curent de la un curent de cationi conductori de ioni, este un curent activat de hiperpolarizare a cationilor monovalenți prin intermediul porilor de închidere S4 care contracarează curenții de rectificare K+) depolarizând sau destabilizând potențialul de repaus.
segmentul S4 se deplasează spre exterior în timpul depolarizării închizând calea conductivă. Fibrele musculare cu mutații severe ale senzorului de tensiune sunt depolarizate nu numai în timpul hipokaliemiei, ci și la nivelurile de potasiu în intervalul normal, explicând slăbiciunea Interictală și permanentă. Miopatia severă cu înlocuirea grasă a țesutului muscular se găsește frecvent la pacienții cu Cav1.1 R1239H (mutații DIV).
glucocorticosteroizii determină Hipopp prin stimularea Na+ K+ ATPază mediată de insulină și amilină.
Gena canalului de potasiu
rectificarea interioară este o proprietate importantă a canalelor Kir. Rectificarea implică blocarea porilor de conducere dependentă de tensiune a porilor cu poliamine și Mg++ în timpul depolarizării, iar acest blocaj este îndepărtat în timpul gradientului potențial în timpul hiperpolarizării. Mutațiile canalului de potasiu sunt observate în sindromul Andersen-Tawil și PP tirotoxic.
triada caracteristicilor dismorfice, paralizia periodică și aritmiile cardiace caracterizează sindromul Andersen-Tawil. Acest sindrom este asociat cu mutații ale genei KCNJ2. Gena KCNJ2 codifică canalul de potasiu rectificator interior Kir2.1. S-a raportat că mutațiile canalului de potasiu în KCNE3 provoacă PP hipokalemic, dar acest lucru nu a fost justificat.
mutațiile din Kir2.6 determină susceptibilitatea la PP tirotoxic. Slăbiciunea episodică observată în PP tirotoxic este similară cu cea observată în Hipopp și sindromul Andersen-Tawil. Această tulburare este cea mai răspândită la bărbații asiatici și latino-americani. PP tirotoxic este o tulburare genetică demascată de tirotoxicoză. Kir2.6 este exprimat în principal în mușchii scheletici. Triiodotironina îmbunătățește transcrierea KCNJ18, care poate conduce la o expresie îmbunătățită a Kir2.6. PKC este activat în timpul tirotoxicozei din cauza creșterii cifrei de afaceri a PIP2, iar canalele Kir interacționează direct cu PIP2 în timpul închiderii normale. În sindromul Andersen-Tawil, există o afinitate scăzută a PIP2. În PP tirotoxic, niciuna dintre mutații nu modifică rectificarea Kir2.6.