Sindromul Aicardi-gouti Otrivres: spectru fenotipic și genetic într-o serie de trei cazuri | Anales de Pediatr Oqua
sindromul Aicardi-gouti Otrivres (AGS) este o boală ereditară rară a cărei prevalență exactă nu este cunoscută. A fost descrisă pentru prima dată în 1984 de Jean Aicardi și Francoise gouti Otrivres ca o encefalopatie progresivă cu debut în primele luni de viață caracterizată prin limfocitoză cu lichid cefalorahidian (LCR) și calcificări în ganglionii bazali.1 se manifestă prin iritabilitate, retard psihomotor, spasticitate, distonie, convulsii epileptice, episoade recurente de febră aseptică și microcefalie. Mortalitatea este mai mare în timpul fazei encefalopatice și, deși boala se stabilizează de obicei după aceea, provoacă sechele neurologice severe. Alte caracteristici caracteristice care pot apărea în cursul acestuia sunt chilblains, simptome oculare (în principal glaucom), afectare cardiacă sau tulburări autoimune.2 interferonii de tip I joacă un rol crucial în patogeneza AGS, în care expresia lor este reglată în sus, ceea ce duce la creșterea producției.3 din acest motiv, una dintre descoperirile clasice de laborator la acești pacienți este un nivel crescut de interferon alfa în LCR, împreună cu pleocitoza și niveluri la fel de ridicate de neopterină și biopterină. Utilitatea potențială a evaluării nivelului de exprimare a genelor stimulate de interferon de către interferon în sângele periferic ca marker este în prezent investigată, deoarece există dovezi că aceste niveluri rămân ridicate după faza encefalopatică („semnătura interferonului”).3-5 o altă caracteristică cheie este detectarea anomaliilor neuroimagistice, inclusiv calcificări în ganglionii bazali și modificări ale substanței albe (Fig. 1). Până în prezent, cunoaștem 7 gene ale căror mutații pot duce la reglarea în sus a căii interferonului: ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1 și IFIH1. Mutațiile heterozigote au fost descrise pentru genele TREX1, ADAR și IFIH1, în timp ce mutațiile raportate în toate celelalte gene au fost homozigote.2 mutațiile genei IFIH1 au fost detectate cel mai recent (2014)4 și, prin urmare, sunt cele mai puțin frecvente variante patogene, în timp ce mutațiile genelor RNASEH2B și TREX1 reprezintă cea mai mare proporție de cazuri diagnosticate de AGS.
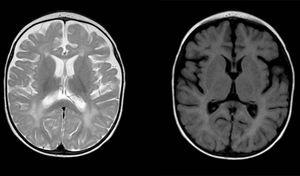
anomalii ale semnalului difuz și neuniform în materia albă în ambele emisfere cerebrale, hiperintense în imaginile ponderate T2. Spațiul subarahnoid mărit cu predominanță frontotemporală în ambele emisfere, cu lărgirea fisurii interhemisferice și creșterea dimensiunii ventriculare (în absența presiunii crescute), compatibilă cu atrofia corticală și subcorticală.
în ultimele decenii, datorită progreselor în genetică care permit detectarea acestor mutații specifice, au apărut dovezi ale unui spectru fenotipic larg dincolo de prezentarea clasică bazată pe gena cauzală. Prezentăm cazurile a 3 pacienți diagnosticați cu AGS în ultimii 8 ani cu scopul de a analiza caracteristicile lor clinice în raport cu defectul genetic de bază (Tabelul 1). În general, caracteristicile prezente ale AGS au fost în concordanță cu cele descrise în cele mai recente serii de cazuri din literatură: prezentare neonatală (33%), microcefalie (66%), retard psihomotor (100%), spasticitate (100%), dizabilitate intelectuală severă (66%) și calcificări pe CT cranian (66%), deși un singur pacient a avut convulsii epileptice.
caracteristicile pacienților cu sindrom Aicardi-gouti Otrivres.
| cazul 1 | Cazul 2 | Cazul 3 | |
|---|---|---|---|
| genetică | mutație homozigotă (p.Ala177Thr) în gena RNASEH2B | mutație homozigotă (341g>A) în gena TREX1 | mutație heterozigotă (c.992C>G și p.Thr331Arg) în gena IFIH1 |
| vârsta curentă | 3 ani | 7 ani și 4 luni | 12 ani și 11 luni |
| Sex | bărbat | femeie | bărbat |
| origine | România | Spania | Italia |
| AP | – | săptămâna 36: restricție de creștere intrauterină săptămâna 37: microcefalie, calcifiere placentară |
palat despicat |
| manifestări clinice | |||
| vârsta la debut | 10 luni | naștere | 2 ani |
| prezentare inițială | iritabilitate regresie psihomotorie |
tremor, hipotonie, plâns slab, eșec de creștere | întârziere a abilităților motorii |
| retard psihomotor | Da | Da | Da |
| limba | 2-silabă words | No | Yes |
| Microcephaly | Yes | Yes | No |
| Chilblains | No | Yes | Yes |
| Epilepsy | No | Yes | No |
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFCS IV |
| tulburări de mișcare | nu | Da | nu |
| mișcări anormale ale ochilor | nu | nu | nu |
| tulburări vizuale | nu | – | miopie |
| glaucom | nu | nu | nu |
| pierderea auzului | – | – | nu |
| afectare cardiacă | No | regurgitare ușoară tricuspidă și mitrală | nu |
| febră recurentă | nu | nu | nu |
| dizabilitate intelectuală | Da | da, gravă | Da, ușoară |
| altele | – | – | sindromul Singleton-Merten: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
| CSF | |||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – |
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – |
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – |
| INF alfa (IU/mL) | 25 ( | 75 ( | – |
| Blood | |||
| Interferon signature | – | – | Gene overexpression |
| Neuroimaging | |||
| Calcifications on CT | No | Yes. În ganglionii bazali și periventriculari | Da. Calcificări simetrice în WM profund al regiunii frontale și al nucleului lentiform |
| RMN-ul capului | modificări difuze și neuniforme ale intensității WM a ambelor emisfere cerebrale, hiperintense pe T2. Implicarea WM subcorticală (care economisesc fibrele U) și WM periventriculară | implicarea WM generalizată cu predominanța WM lobară, inclusiv fibrele u subcorticale ale lobilor frontali, temporali și occipitali, bilateral și simetric, fără implicare corticală | – |
CSF, lichid cefalorahidian; CT, tomografie computerizată; GMFC, sistem de clasificare a funcției motorii brute; INF, interferon; IUGR, restricție de creștere intrauterină; RMN, imagistică prin rezonanță magnetică; PNP, polineuropatie; WM, alb mater.
după cum sa menționat anterior, mutațiile homozigote din gena RNASEH2B sunt cele mai frecvente variante care provoacă AGS, iar expresia lor fenotipică se conformează cel mai mult prezentării clasice.4 acesta a fost cazul pacientului din studiul nostru care a purtat o astfel de mutație, care a debutat la vârsta de 10 luni cu iritabilitate și retard psihomotor și cu constatări caracteristice de neuroimagistică și LCR.
douăzeci la sută din cazurile de AGS pot avea o prezentare neonatală, cu debutul bolii care apare in utero.5 mutații în oricare dintre cele 7 gene menționate mai sus pot duce la acest fenotip, dar această prezentare timpurie este cel mai frecvent asociată cu gena TREX.4,5 prezentarea inițială a acestei forme este similară cu cea a unei infecții TORCH, cu hepatosplenomegalie, hipertransaminazemie, trombocitopenie și manifestări neurologice incluzând iritabilitate extremă, tulburări de mișcare și crize epileptice.5 acești pacienți au o evoluție mai severă a bolii și prezintă un risc mai mare de deces. Pacientul din eșantionul nostru care a prezentat o astfel de variantă a avut o prezentare neonatală și are în prezent cea mai severă formă de boală a celor 3.
mutațiile genei ADAR1 și în special a genei IFIH1 sunt asociate cu un debut tardiv al simptomelor, după 1 an de viață cu dezvoltare psihomotorie normală.5 în unele dintre aceste cazuri, sindromul are un curs benign, cu păstrarea relativă a limbajului și a abilităților motorii. Pacientul nostru cu o mutație a genei IFIH1 a fost un caz singular prin faptul că a avut și sindromul Singleton-Merten, o boală rară cauzată și de o mutație a genei IFIH1 și caracterizată prin displazie dentară, calcificări aortice și osteoporoză.6
scopul nostru este de a sublinia variabilitatea fenotipică semnificativă a AGS și asocierea sa cu mutații specifice atât în scopul încurajării examinării acestui diagnostic în cazurile cu prezentări care se abat de la forma clasică a bolii, cât și pentru a contribui la informații suplimentare despre evoluția bolii și rezultatele la acești pacienți.