Aicardi-Gouti Aucrires syndrom: fenotypiskt och genetiskt spektrum i en serie av tre fall | Anales de Pediatric Aucarica
Aicardi-Gouti Aucrires syndrom (AGS) är en sällsynt ärftlig sjukdom vars exakta prevalens är okänd. Det beskrevs första gången 1984 av Jean Aicardi och Francoise Gouti Exceprires som en progressiv encefalopati med början under de första månaderna av livet som kännetecknas av cerebrospinalvätska (CSF) lymfocytos och förkalkningar i basala ganglier.1 det manifesterar sig med irritabilitet, psykomotorisk retardation, spasticitet, dystoni, epileptiska anfall, återkommande episoder av aseptisk feber och mikrocefali. Dödligheten är högre under den encefalopatiska fasen, och även om sjukdomen vanligtvis stabiliseras efteråt orsakar den allvarliga neurologiska följder. Andra karakteristiska egenskaper som kan uppstå under kursen är kylskador, okulära symtom (främst glaukom), hjärtinvolvering eller autoimmuna störningar.2 typ i-interferoner spelar en avgörande roll i patogenesen av AGS, där deras uttryck uppregleras vilket leder till ökad produktion.3 av denna anledning är ett av de klassiska laboratoriefynden hos dessa patienter en förhöjd nivå av interferon alfa i CSF, tillsammans med pleocytos och lika förhöjda nivåer av neopterin och biopterin. Den potentiella nyttan av att bedöma expressionsnivån av interferonstimulerade gener av interferon i perifert blod som markör undersöks för närvarande, eftersom det finns bevis för att dessa nivåer förblir höga förbi den encefalopatiska fasen (”interferonsignatur”).3-5 en annan viktig funktion är detektering av neuroimaging abnormiteter inklusive förkalkningar i basala ganglier och förändringar i den vita substansen (Fig. 1). Hittills känner vi till 7 gener vars mutationer kan leda till uppreglering av interferonvägen: ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1 och IFIH1. Heterozygota mutationer har beskrivits för trex1 -, ADAR-och ifih1-generna, medan mutationerna som rapporterats i alla andra gener har varit homozygota.2 mutationer i ifih1-genen upptäcktes senast (2014) 4 och är därför de minst frekventa patogena varianterna, medan mutationer i rnaseh2b-och TREX1-generna står för den högsta andelen diagnostiserade fall av AGS.
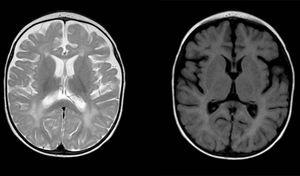
diffusa och fläckiga signalavvikelser i vit materia i båda hjärnhalvorna, hyperintensitet i T2-viktade bilder. Förstorat subaraknoidutrymme med frontotemporal övervägande i båda halvkärmen, med utvidgning av den interhemisfäriska sprickan och ökad ventrikulär storlek (i frånvaro av ökat tryck), kompatibel med kortikal och subkortisk atrofi.
under de senaste decennierna, tack vare framsteg inom genetik som möjliggör detektering av dessa specifika mutationer, har bevis framkommit av ett brett fenotypiskt spektrum utöver den klassiska presentationen baserad på den orsakande genen. Vi presenterar fallen av 3 patienter som fått en diagnos av AGS under de senaste 8 åren i syfte att analysera deras kliniska egenskaper i förhållande till den underliggande genetiska defekten (Tabell 1). I allmänhet överensstämde de presenterande egenskaperna hos AGS med de som beskrivs i den senaste fallserien i litteraturen: neonatal presentation (33%), mikrocefali (66%), psykomotorisk retardation (100%), spasticitet (100%), svår intellektuell funktionsnedsättning (66%) och förkalkningar på kranial CT (66%), även om endast en patient hade epileptiska anfall.
egenskaper hos patienter med Aicardi-Gouti-syndrom.
| fall 1 | Fall 2 | fall 3 | |
|---|---|---|---|
| genetik | homozygot mutation (p. Ala177Thr) i rnaseh2b gen | homozygot mutation (341G>a) i trex1 gen | heterozygot mutation (c.992C>G och p.Thr331Arg) i ifih1-genen |
| nuvarande ålder | 3 år | 7 år och 4 månader | 12 år och 11 månader |
| kön | hane | Kvinna | hane |
| Ursprung | Rumänien | Spanien | Italien |
| AP | – | Vecka 36: intrauterin tillväxtbegränsning Vecka 37: mikrocefali, placenta förkalkning |
gomspalt |
| kliniska manifestationer | |||
| ålder vid debut | 10 månader | födelse | 2 år |
| inledande presentation | irritabilitet psykomotorisk regression |
skakningar, hypotoni, svag gråt, tillväxtfel | fördröjning av motorisk skicklighet |
| psykomotorisk retardation | Ja | Ja | Ja |
| språk | 2-stavelse words | No | Yes |
| Microcephaly | Yes | Yes | No |
| Chilblains | No | Yes | Yes |
| Epilepsy | No | Yes | No |
| Motor impairment | Spastic tetraplegia. GMFCS IV | Severe spastic tetraplegia. GMFCS V | Spastic tetraplegia. GMFCS IV |
| rörelsestörning | Nej | Ja | Nej |
| onormala ögonrörelser | Nej | Nej | Nej |
| synnedsättning | Nej | – | myopi |
| glaukom | Nej | Nej | Nej |
| hörselnedsättning | – | – | Nej |
| Hjärtinvolvering | Nej | Mild tricuspid och mitral regurgitation | Nej |
| återkommande feber | Nej | Nej | Nej |
| intellektuell funktionsnedsättning | Ja | ja, grav | ja, mild |
| Övriga | – | – | Singleton-Merten syndrom: tooth abnormalities Axonal and demyelinating sensorimotor PNP |
| CSF | |||
| CSF analysis | Increased protein | Pleocytosis and increased protein | – |
| Neopterin (nmol/L) | 1.102 (12–55) | 1.813 (12–55) | – |
| Biopterin (nmol/L) | 66 (22–73) | 53 (22–73) | – |
| INF alfa (IU/mL) | 25 ( | 75 ( | – |
| Blood | |||
| Interferon signature | – | – | Gene overexpression |
| Neuroimaging | |||
| Calcifications on CT | No | Yes. I basala och periventrikulära ganglier | Ja. Symmetriska förkalkningar i djup WM av frontal region och lentiform kärna |
| Huvud Mr | diffusa och fläckiga förändringar i WM-intensiteten hos båda hjärnhalvorna, hyperintensitet på T2. Involvering av subkortisk WM (skonar u-fibrerna) och periventrikulär WM | generaliserat WM-engagemang med övervägande av lobar WM inklusive de subkortiska u-fibrerna i frontala, temporala och occipitala lober, bilateralt och symmetriskt, utan kortikal involvering | – |
CSF, cerebrospinalvätska; CT, datortomografi; GMFC, klassificeringssystem för grovmotorisk funktion; INF, interferon; IUGR, intrauterin tillväxtbegränsning; Mr, magnetisk resonansavbildning; PNP, polyneuropati; WM, vit mater.
som nämnts tidigare är homozygota mutationer i rnaseh2b-genen de vanligaste varianterna som orsakar AGS och deras fenotypiska uttryck överensstämmer vanligtvis mest med den klassiska presentationen.4 Detta var fallet med patienten i vår studie som bar en sådan mutation, som hade börjat vid 10 års ålder med irritabilitet och psykomotorisk retardation och med karakteristiska neuroimaging-och CSF-fynd.
tjugo procent av fallen av AGS kan ha en neonatal presentation, med sjukdomsuppkomsten i utero.5 mutationer i någon av de 7 ovannämnda generna kan leda till denna fenotyp, men denna tidiga presentation är oftast associerad med Trex-genen.4,5 den initiala presentationen av denna form liknar den för en TORCH-infektion, med hepatosplenomegali, hypertransaminasemi, trombocytopeni och neurologiska manifestationer inklusive extrem irritabilitet, rörelsestörningar och epileptiska anfall.5 Dessa patienter har en allvarligare sjukdomsförlopp och har högre risk för dödsfall. Patienten i vårt prov som presenterades med en sådan variant hade en neonatal presentation och har för närvarande den allvarligaste sjukdomsformen hos 3.
mutationer i ADAR1-genen och särskilt ifih1-genen är associerade med en sen symtomdebut efter 1 levnadsår med normal psykomotorisk utveckling.5 i vissa av dessa fall har syndromet en godartad kurs med relativ bevarande av språk och motoriska färdigheter. Vår patient med en mutation i ifih1-genen var ett singulärt fall genom att han också hade Singleton – Merten syndrom, en sällsynt sjukdom som också orsakades av en mutation i ifih1-genen och kännetecknades av dental dysplasi, aortaförkalkningar och osteoporos.6
vårt mål är att understryka den signifikanta fenotypiska variationen hos AGS och dess samband med specifika mutationer i syfte att både uppmuntra övervägande av denna diagnos i fall med presentationer som avviker från den klassiska sjukdomsformen och att bidra med ytterligare information om sjukdomsförloppet och resultaten hos dessa patienter.