gynekologi och obstetrik fallrapport
nyckelord
Tetra-amelia; missbildning; genotyp; fenotyp.
introduktion
lem anomalier utgör en viktig grupp av medfödda patologier som kännetecknas av hypoplasi eller fullständig aplasi av en eller flera benben. Lemmavvikelser av alla typer förekommer hos cirka 1 av 1 300 till 2 000 födda. Dessa anomalier i benen kan isoleras eller associeras med andra anomalier . Tetraamelia syndrom är sällsynt och gråa områden kvarstår.
vi rapporterar två fall av tetra-amelia i en nivå II Moderskap i Dakar (Senegal) med att likna tetraamelia-1 (kromosom 17q21), tetraamelia-2 (kromosom 8q23) och Roberts syndrom (kromosom 8p21). Detta illustrerar svårigheten att korrelera fenotyp och involverade gener.
fallrapporter
fall 1
ms AD var en 44-årig mamma som hänvisades till vår avdelning vid 36 veckors graviditet med svår preeklampsi och fosteranomalier. Hon var fem para utan historia av fosteranomalier. Hon rökte inte nu och hade aldrig varken rökt eller druckit alkohol. Hon hade inte blivit utsatt för passiv rökning. Hon var i en tredje gradens blodsförvanter äktenskap för alla sina barn. Ms AD hade testat negativt för hepatit B, HIV och syfilis. Hon var skyddad från rubellaviruset och hade ingen tidigare exponering för Toxoplasma gondii. Ultraljudsövervakning utfördes sent vid 33 veckor och 35 veckors graviditet hämtad oligoamnios och hydrocephalus samt agenes i lemmarna. Recept under graviditeten inkluderade administrering av järn och folsyra samt administrering av sulfadoxinpyrimetamin. Den senare ordinerades vid 18 veckor och sedan 26 veckor som en del av anti-malariaprofylaxpolitiken för gravida kvinnor. Symphyseal-fundal höjd mätt 28 cm. På grund av svåra egenskaper hos preeklampsi; hon var omedelbart inlagd på sjukhus och observerades i en arbets-och leveransenhet. Hon fick sedan initialt IV magnesiumsulfat för att förhindra eklampsi och antihypertensiva läkemedel för att upprätthålla systoliskt blodtryck under 160 mmHg och diastoliskt blodtryck under 105 mmHg.
beslutet om omedelbar kejsarsnitt gjordes. En 2,150 gram levande född extraherades som därefter dog inom 10 minuter. Före döden presenterade kroppen krypande rörelser. Flera externa anomalier identifierades (Figur 1) inklusive fullständig agenes av alla fyra lemmar, hydrocephalus med en huvudomkrets på 39 cm. På ansiktet fanns hypertelorism med ögonlockens kolobom, milda exoftalmos och aniridia. Munnen var lika en inverterad V utan tydlig avgränsning av läpparna och näsan var rudimentär. Den nyfödda saknade integritet (hår och ögonbryn). Öronen reducerades till skisser som såg ut som slitsar. Nacken var kort. Stammen reducerades till en 26 cm lång konisk struktur med en navelsträng i bottenänden. Bröstet var smalt. Strax under naveln var stammen vidgad bakåt närvarande vid golvet i den kaudala Polen, en mittlinje med recessioner och en spirande som kan motsvara en fallus av obestämd typ. Agenes av bäcken, könsorgan och anal ofullkomlighet noterades. Fosterpatologi har inte utförts. Döden inom 10 minuter efter leverans och bröstets koniska utseende kan dock föreslå lungavvikelser.
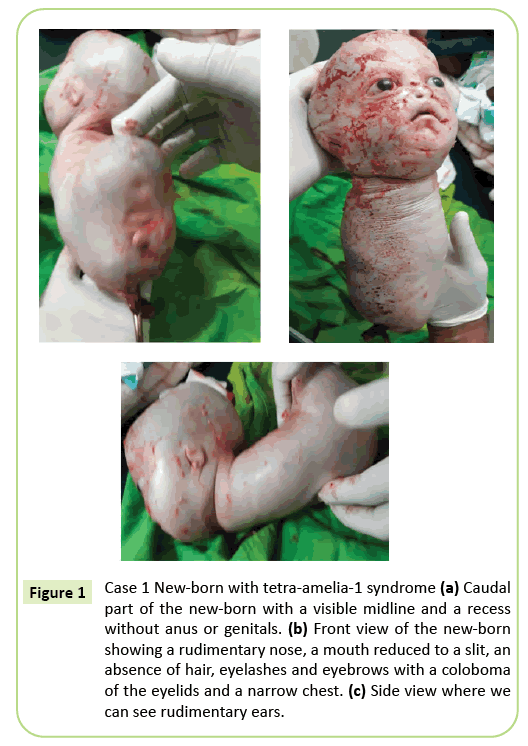
Figur 1: Fall 1 nyfödd med Tetra-amelia-1-syndrom (a) kaudal del av den nyfödda med en synlig mittlinje och en urtagning utan anus eller könsorgan. b) framifrån av den nyfödda som visar en rudimentär näsa, en mun reducerad till en slits, en frånvaro av hår, ögonfransar och ögonbryn med ögonlockens kolobom och ett smalt Bröst. (C) sidovy där vi kan se rudimentära öron.
Fall 2
det andra fallet var en 22-årig primigravida hänvisade till vår anläggning för ultraljudsskanning vid 37 veckors graviditet. Hon var inte i ett konsanguineous äktenskap. Hon hade testat negativt för hepatit B, HIV och syfilis. Hon testades inte för toxoplasmos och rubella. Ingen ultraljudsövervakning gjordes under graviditeten. Den kliniska undersökningen överensstämde med fostrets tillväxtnedgång (fondhöjd: 26 cm). Ultraljudsfynd visade att humerus var förvrängd och mätte 23,9 mm motsvarande 17 veckors graviditet. Det fanns agenes av lårbenet. Iliacvingarna var synliga på ultraljud. Inga lung-eller hjärtavvikelser identifierades. Leverans inleddes. Den nyfödda hade en kvinnlig fenotyp med en Apgar-poäng på 9 vid 5: e minuten. Morfologin hos huvudet och stammen var utan särdrag. De övre extremiteterna reducerades till två 3 cm långa stubbar. Fullständig agenes av de 2 nedre extremiteterna noterades. Det var en symmetrisk anomali (Figur 2).
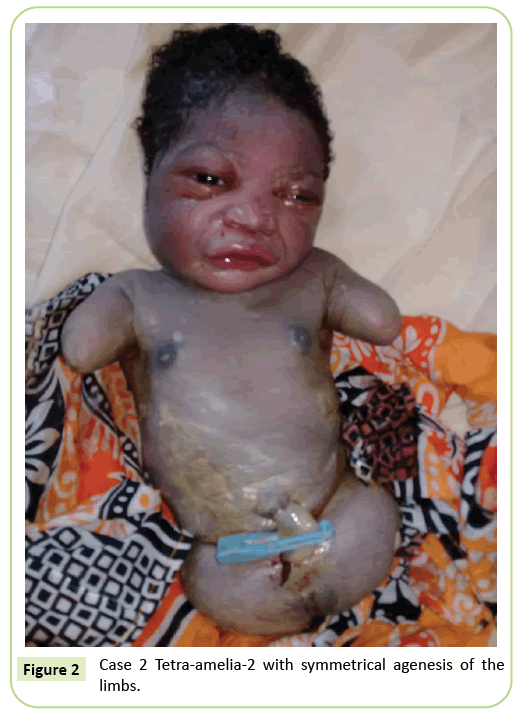
Figur 2: Fall 2 Tetra-amelia-2 med symmetrisk agenes av lemmarna.
diskussion
Bermejo-Sanchez et al. beskrivs i 2011 epidemiologin av medfödd amelia med hjälp av data som samlats in från 20 medfödda anomalier övervakningsprogram, från alla kontinenter utom Afrika, mellan 1968 och 2006. Totalt identifierades 326 fall av amelia bland 23 110 591 levande födda, dödfödda och aborter. Prevalensen var 1,41 / 100 000 .
Tetra-amelia hänvisar till som fullständig frånvaro av lemmarna och förekommer mer sällan. Så vitt vi vet beskrivs tetra-amelia-1 i 7 familjer. Det verkar följa ett autosomalt recessivt arv. I alla familjer var tetra-amelia-1 associerad med allvarliga missbildningar av andra delar av kroppen inklusive ansikte och huvud, anomalier i nervsystemet, skelett och könsorgan. Lungorna var underutvecklade i många fall, vilket gör andningen svår eller omöjlig . Zimmer et al. rapporterade 1985 en starkt inavlad familj där 6 spädbarn hade tetra-amelia-1 och hydrocephalus. De beskrev i ett av fostren en total frånvaro av bäckenben, klyftläpp och gom, arrhinia och aplasi i öronen. En bilateral vänster lunga, en ihållande arteriell kanal, en anal ofullkomlighet noterades också. Fostertestning eliminerade diagnosen Roberts syndrom . Andra fall som hämtats i litteraturen inkluderar Kosaki et al., 1996, med ett foster av karyotyp 46, XX med tetraphocomelia och svår lunghypoplasi förutom ansikts-och huvudanomalier . Rosenak et al. beskrev ett fall av tetra-amelia med svår lunghypoplasi hos två foster av ett icke-konsanguineous par. Fostertestning utesluter diagnos av Roberts syndrom . Ytterligare två fall rapporterades av Zlotogora et al. 1993. Båda patienterna dog strax efter födseln och författarna föreslog förekomsten av lunghypoplasi. Niemann et al. rapporterade en konsanguineous Turkisk familj där 4 av de 8 bröderna led av tetra-amelia. Förutom frånvaron av de 4 lemmarna avslöjade fosterundersökningarna av 3 Foster flera anomalier: kluvna läppar och /eller palatin, laparoschisis, lunganomalier, bäckenets hypoplasi, choanas atresi, vagina och anal ofullkomlighet . Slutligen, 2005, Krahn et al. beskrev 2 bröder födda till inavlade föräldrar som lider av tetraamelia och svår lunghypoplasi. Kragebenen och axelbladen var normala i det andra fostret. Karyotypen var normal .
Tetra-amelia-1-syndrom eller TETAMS1 orsakas av en homozygot mutation i wnt3-genen på kromosom 17q21 med en autosomal recessiv arv. Tetraamelia-2-syndrom (TETAMS2) kännetecknas av rudimentära lemmar eller en fullständig frånvaro av lemmarna, i allmänhet symmetrisk såväl som bilateral agenes i lungorna i vissa fall. Är också vanliga anomalier i lungvaskulärsystemet och dysmorfier inklusive bilateral klyftläpp och gom, ankyloglossi, mandibulär hypoplasi, mikroretrognathia och labioscrotal aplasi .
Szenker-Ravi, som studerade 4 familjer av tetra-amelia med agenes eller lunghypoplasi, noterade en fenotypisk heterogenitet med lemmavvikelser av varierande svårighetsgrad . Exome-sekvensering i dessa 4-familjer har gjort det möjligt att identifiera trunkerande homozygota mutationer i rspo2-genen . Tetraamelia-2-syndrom orsakas av en homozygot mutation i rspo2-genen (610575) belägen på kromosom 8q23 .
fenotypen av det första fallet som beskrivs i denna artikel motsvarar ett tetra-amelia-1-syndrom, särskilt på grund av närvaron av hydrocephalus, anomalier könsorgan och en rudimentär näsa. Det smala bröstet och den tidiga döden före den 10: e minuten av livet tyder på allvarlig lunghypoplasi. Detta fall belyser den fenotypiska heterogeniteten med ett ögonlockskolobom, hypertelorism, exoftalmos och sällsynta bilagor.
vi anser att det andra fallet i vår studie är ett tetramelia-2-syndrom med tanke på den symmetriska tetra-amelia med närvaron av övre extremiteter. Diagnos av tetra-amelia bör göras tidigt under ultraljudsövervakning. Därför bör medvetenheten höjas om vikten av ultraljudsövervakning och användningen av 3D/4D för att förbättra screeningsresultaten. Diagnosen av en bäckenmassa på ultraljud ihop med amelia bör väcka misstankar för spleno-gonadal fusion lem defekt syndrom.
dessutom ska fosterundersökningen och fostertestningen med hjälp av den utvecklande tekniken för kromosomal mikroarray och exome och genomsekvensering uppmuntras i våra inställningar. En bättre karaktärisering av fallen gör det möjligt att ge råd till par och en bättre kunskap om dessa kliniska avvikelser.
slutsats
Tetra-amelia syndrom är knappt och gråa områden kvarstår. Dessa två fall, jämfört med vad som redan beskrivits i litteraturen, illustrerar den fenotypiska heterogeniteten hos tetraamelia. Med tanke på den sällsynta förekomsten av dessa anomalier skulle det vara viktigt att skapa ett internationellt register över anomalier för att rapportera fall och inrätta en provbank för utökade genetiska studier till föräldrar.
- Wilcox WR, Coulter CP, Schmitz ML (2015) medfödda lembriststörningar. Clin Perinatol 42: 281-300.
- Bermejo SE, Cuevas L, Amar E (2011) Amelia: en multi-center beskrivande epidemiologisk studie i en stor dataset från International Clearinghouse for Birth Defects Surveillance and Research, och översikt över litteraturen. Am J Med Genet C Semin Med Genet 157: 288-304.
- Zlotogora JSM, Shabany YO, Jarallah RY (1993) syndrom av tetraamelia med lunghypoplasi. Am J Med Genet 47: 570-571.
- Zimmer EZ (1985) Tetra-amelia med flera missbildningar hos sex manliga foster i en släkt. Europ. J Pediat 144: 412-414.
- Rosenak D (1991) återkommande tetraamelia och lunghypoplasi med flera missbildningar i sibs. Am J Med Genet 38: 25-28.
- Gershoni BR (1990) Roberts syndrom eller ”X-länkad amelia” ? . Am J Med Genet 37: 569-572.
- Kosaki K, Jones MC, Stayboldt C (1996) Zimmer phocomelia: avgränsning genom huvudkoordinatanalys. Am J Med Genet 66: 55-59.
- Niemann S(2004) homozygot wnt3-mutation orsakar tetra-amelia i en stor konsanguineous familj. Am J Hum Genet 74: 558-563.
- Krahn M (2005)Tetra-amelia och lung aplasia syndrom: rapport om en ny familj och uteslutning av kandidatgener. Clin Genet 68: 558-560.
- Szenker-RE, Altunoglu U (2018) RSPO2-hämning av RNF43 och ZNRF3 styr lemmutveckling oberoende av LGR4/5/6. Natur 557: 564-569.