MO för HF
molekylära orbitaler för heterogena diatomiska molekyler
en enkel metod för molekylär orbital (MO) teori för heterogena diatomiska molekyler är att visa energinivådiagrammet. MO – energinivåerna kan utarbetas enligt dessa steg:
minns att energin \(E_n\) för kvantnumret n är för ett element med Atom Z är ungefär
\
vi använder \(Z_{eff}\) istället för Z för att betyda att vi måste ändra atomnumret för att få en effektiv atomladdning för kärnan. Eftersom vi har att göra med ungefärliga värden kan man använda Z direkt. 1s-orbitalenerginivån är -13,6 eV för väteatomer, mätt som joniseringsenergin för H.
således för kvantnumret n = 1 är energinivån för 1s av He ungefär – 54 eV. På samma sätt är 1s energinivån för F – 1101 eV. 2s – och 2P-energinivåerna för He är ungefär-13.6 eV, vilket är simlar till 1s-orbitalen av H.
således är 2s-energinivån för Li ungefär -6 eV. Men för multielektronatomer har p-subshell och s-subshell olika energier på grund av penetration. På den här nivån kan vi inte vara exakta om det, utan bara tro att 2P-orbitalerna har högre energi än 2s-orbitalen. Vanligtvis överlappar atomorbitaler med energinivåer som liknar varandra för att bilda molekylära orbitaler. Således matchar vi energinivåerna hos atomorbitaler och gör sedan bindning och antibindning av dem.
men om den atomära orbitalenerginivån är väldigt annorlunda använder vi atomorbitaler i det ofullständiga delskalet för att bilda MOs.
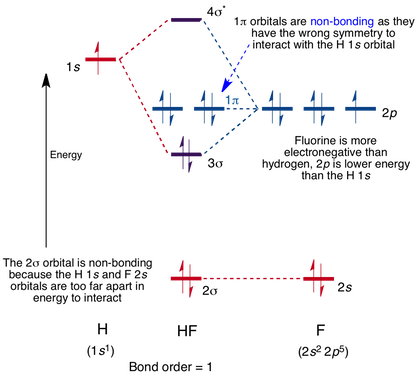
molekylärt Orbitaldiagram för HF-molekylen
interaktion sker mellan 1s-orbitalen på väte och 2P-orbitalen i fluor som orsakar bildandet av en sigma-bindning och en Sigma-antibonding molekylär orbital, som visas nedan.